
Impact of Fecal Microbiota Transplantation on Gut Bacterial Bile Acid Metabolism in Humans

 ,
,  , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Microbiota Analysis
2.3. Data Analysis and Statistics
3. Results
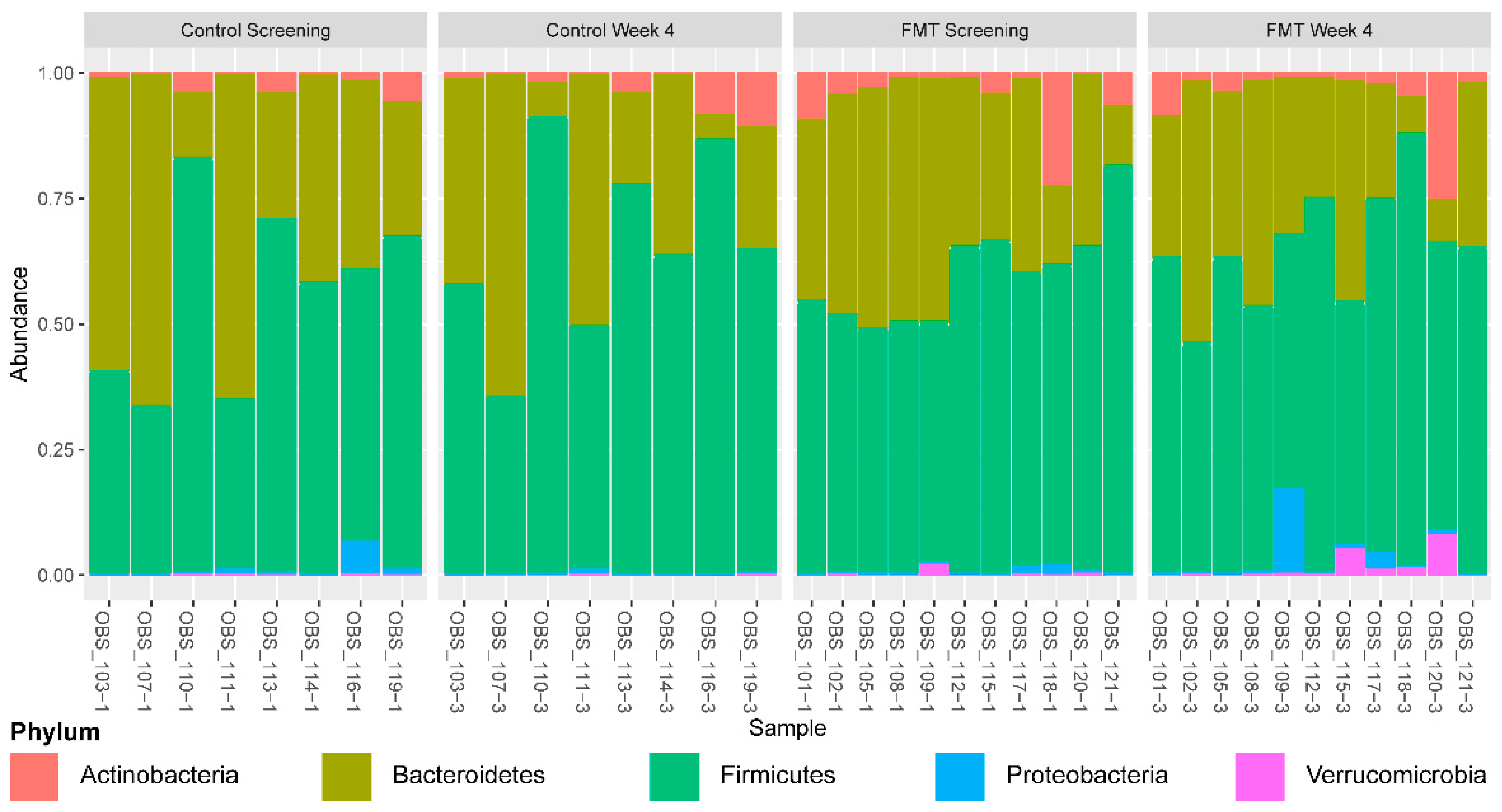
3.1. Microbial Diversity
3.2. Impact of FMT on Gut Microbial Composition at 4 Weeks after the Initiation of Intervention
3.3. Impact of FMT on Gene Enrichment and Correlations with Secondary Bile Acid Production
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut Metagenome in European Women with Normal, Impaired and Diabetic Glucose Control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A Core Gut Microbiome in Obese and Lean Twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human Gut Microbes Associated with Obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Backhed, F.; Fulton, L.; Gordon, J.I. Marked Alterations in the Distal Gut Microbiome Linked to Diet-Induced Obesity. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marotz, C.A.; Zarrinpar, A. Treating Obesity and Metabolic Syndrome with Fecal Microbiota Transplantation. Yale J. Biol. Med. 2016, 89, 383–388. [Google Scholar]
- Kassam, Z.; Lee, C.H.; Yuan, Y.; Hunt, R.H. Fecal Microbiota Transplantation for Clostridium Difficile Infection: Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2013, 108, 500–508. [Google Scholar] [CrossRef]
- Hourigan, S.K.; Ahn, M.; Gibson, K.M.; Pérez-Losada, M.; Felix, G.; Weidner, M.; Leibowitz, I.; Niederhuber, J.E.; Sears, C.L.; Crandall, K.A.; et al. Fecal Transplant in Children with Clostridioides Difficile Gives Sustained Reduction in Antimicrobial Resistance and Potential Pathogen Burden. Open Forum Infect. Dis. 2019, 6, ofz379. [Google Scholar] [CrossRef] [Green Version]
- Allegretti, J.R.; Kassam, Z.; Hurtado, J.; Marchesi, J.R.; Mullish, B.H.; Chiang, A.; Thompson, C.C.; Cummings, B.P. Impact of Fecal Microbiota Transplantation with Capsules on the Prevention of Metabolic Syndrome among Patients with Obesity. Hormones 2021, 20, 209–211. [Google Scholar] [CrossRef]
- Allegretti, J.R.; Kassam, Z.; Mullish, B.H.; Chiang, A.; Carrellas, M.; Hurtado, J.; Marchesi, J.R.; McDonald, J.A.K.; Pechlivanis, A.; Barker, G.F.; et al. Effects of Fecal Microbiota Transplantation with Oral Capsules in Obese Patients. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2020, 18, 855–863.e2. [Google Scholar] [CrossRef]
- Hamilton, J.P.; Xie, G.; Raufman, J.-P.; Hogan, S.; Griffin, T.L.; Packard, C.A.; Chatfield, D.A.; Hagey, L.R.; Steinbach, J.H.; Hofmann, A.F. Human Cecal Bile Acids: Concentration and Spectrum. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G256–G263. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G Protein-Coupled Receptor Responsive to Bile Acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a Nuclear Receptor for Bile Acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of Membrane-Type Receptor for Bile Acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Makki, K.; Brolin, H.; Petersen, N.; Henricsson, M.; Christensen, D.P.; Khan, M.T.; Wahlström, A.; Bergh, P.-O.; Tremaroli, V.; Schoonjans, K.; et al. 6α-Hydroxylated Bile Acids Mediate TGR5 Signalling to Improve Glucose Metabolism upon Dietary Fiber Supplementation in Mice. Gut 2022. [Google Scholar] [CrossRef] [PubMed]
- Russell, B.J.; Brown, S.D.; Siguenza, N.; Mai, I.; Saran, A.R.; Lingaraju, A.; Maissy, E.S.; Dantas Machado, A.C.; Pinto, A.F.M.; Sanchez, C.; et al. Intestinal Transgene Delivery with Native E. coli Chassis Allows Persistent Physiological Changes. Cell 2022, 185, 3263–3277.e15. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Bile Salt Biotransformations by Human Intestinal Bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile Acids and the Gut Microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.V.; Begley, M.; Hill, C.; Gahan, C.G.M.; Marchesi, J.R. Functional and Comparative Metagenomic Analysis of Bile Salt Hydrolase Activity in the Human Gut Microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 13580–13585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullish, B.H.; McDonald, J.A.K.; Pechlivanis, A.; Allegretti, J.R.; Kao, D.; Barker, G.F.; Kapila, D.; Petrof, E.O.; Joyce, S.A.; Gahan, C.G.M.; et al. Microbial Bile Salt Hydrolases Mediate the Efficacy of Faecal Microbiota Transplant in the Treatment of Recurrent Clostridioides Difficile Infection. Gut 2019, 68, 1791–1800. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. Babraham Bioinformatics—FastQC a Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 17 October 2022).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Rahnavard, A.; Mann, B.; Giri, A.; Chatterjee, R.; Crandall, K.A. Metabolite, Protein, and Tissue Dysfunction Associated with COVID-19 Disease Severity. Sci. Rep. 2022, 12, 12204. [Google Scholar] [CrossRef] [PubMed]
- Mallick, H.; Chatterjee, S.; Chowdhury, S.; Chatterjee, S.; Rahnavard, A.; Hicks, S.C. Differential Expression of Single-Cell RNA-Seq Data Using Tweedie Models. Stat. Med. 2022, 41, 3492–3510. [Google Scholar] [CrossRef]
- Kitahara, M.; Takamine, F.; Imamura, T.; Benno, Y. Assignment of Eubacterium sp. VPI 12708 and Related Strains with High Bile Acid 7α-Dehydroxylating Activity to Clostridium Scindens and Proposal of Clostridium hylemonae sp. Nov., Isolated from Human Faeces. Int. J. Syst. Evol. Microbiol. 2000, 50 Pt 3, 971–978. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Isolation and Characterization of a Bile Acid Inducible 7α-Dehydroxylating Operon in Clostridium hylemonae TN271. Anaerobe 2010, 16, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, N.; Chen, Q.; Qin, H. Fecal Microbiota Transplantation Modulates the Gut Flora Favoring Patients with Functional Constipation. Front. Microbiol. 2021, 12, 700718. [Google Scholar] [CrossRef]
- Massot-Cladera, M.; Azagra-Boronat, I.; Franch, À.; Castell, M.; Rodríguez-Lagunas, M.J.; Pérez-Cano, F.J. Gut Health-Promoting Benefits of a Dietary Supplement of Vitamins with Inulin and Acacia Fibers in Rats. Nutrients 2020, 12, 2196. [Google Scholar] [CrossRef]
- Pimentel, J.D.; Chan, R.C. Desulfovibrio Fairfieldensis Bacteremia Associated with Choledocholithiasis and Endoscopic Retrograde Cholangiopancreatography. J. Clin. Microbiol. 2007, 45, 2747–2750. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Shao, W.; Liu, Q.; Liu, N.; Wang, Q.; Xu, J.; Zhang, X.; Weng, Z.; Lu, Q.; Jiao, L.; et al. Gut Microbiota Promotes Cholesterol Gallstone Formation by Modulating Bile Acid Composition and Biliary Cholesterol Secretion. Nat. Commun. 2022, 13, 252. [Google Scholar] [CrossRef]
- Van Eldere, J.; Celis, P.; De Pauw, G.; Lesaffre, E.; Eyssen, H. Tauroconjugation of Cholic Acid Stimulates 7α-Dehydroxylation by Fecal Bacteria. Appl. Environ. Microbiol. 1996, 62, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.; Yu, J.; McDowell, A.; Kim, S.H.; You, H.J.; Ko, G. Bile Salt Hydrolase-Mediated Inhibitory Effect of Bacteroides Ovatus on Growth of Clostridium Difficile. J. Microbiol. 2017, 55, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Davis-Richardson, A.G.; Ardissone, A.N.; Dias, R.; Simell, V.; Leonard, M.T.; Kemppainen, K.M.; Drew, J.C.; Schatz, D.; Atkinson, M.A.; Kolaczkowski, B.; et al. Bacteroides Dorei Dominates Gut Microbiome Prior to Autoimmunity in Finnish Children at High Risk for Type 1 Diabetes. Front. Microbiol. 2014, 5, 678. [Google Scholar] [CrossRef] [PubMed]
- Grill, J.P.; Manginot-Dürr, C.; Schneider, F.; Ballongue, J. Bifidobacteria and Probiotic Effects: Action of Bifidobacterium Species on Conjugated Bile Salts. Curr. Microbiol. 1995, 31, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.-B.; Brochet, M.; Lee, B.H. Cloning and Characterization of a Bile Salt Hydrolase (Bsh) from Bifidobacterium adolescentis. Biotechnol. Lett. 2005, 27, 817–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miquel, S.; Leclerc, M.; Martin, R.; Chain, F.; Lenoir, M.; Raguideau, S.; Hudault, S.; Bridonneau, C.; Northen, T.; Bowen, B.; et al. Identification of Metabolic Signatures Linked to Anti-Inflammatory Effects of Faecalibacterium prausnitzii. mBio 2015, 6, e00300-15. [Google Scholar] [CrossRef] [Green Version]
- Quévrain, E.; Maubert, M.A.; Michon, C.; Chain, F.; Marquant, R.; Tailhades, J.; Miquel, S.; Carlier, L.; Bermúdez-Humarán, L.G.; Pigneur, B.; et al. Identification of an Anti-Inflammatory Protein from Faecalibacterium prausnitzii, a Commensal Bacterium Deficient in Crohn’s Disease. Gut 2016, 65, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Hirano, S.; Masuda, N. Characterization of NADP-Dependent 7β-Hydroxysteroid Dehydrogenases from Peptostreptococcus Productus and Eubacterium Aerofaciens. Appl. Environ. Microbiol. 1982, 43, 1057–1063. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bustamante, J.-M.; Dawson, T.; Loeffler, C.; Marfori, Z.; Marchesi, J.R.; Mullish, B.H.; Thompson, C.C.; Crandall, K.A.; Rahnavard, A.; Allegretti, J.R.; et al. Impact of Fecal Microbiota Transplantation on Gut Bacterial Bile Acid Metabolism in Humans. Nutrients 2022, 14, 5200. https://doi.org/10.3390/nu14245200
Bustamante J-M, Dawson T, Loeffler C, Marfori Z, Marchesi JR, Mullish BH, Thompson CC, Crandall KA, Rahnavard A, Allegretti JR, et al. Impact of Fecal Microbiota Transplantation on Gut Bacterial Bile Acid Metabolism in Humans. Nutrients. 2022; 14(24):5200. https://doi.org/10.3390/nu14245200
Chicago/Turabian StyleBustamante, Jessica-Miranda, Tyson Dawson, Caitlin Loeffler, Zara Marfori, Julian R. Marchesi, Benjamin H. Mullish, Christopher C. Thompson, Keith A. Crandall, Ali Rahnavard, Jessica R. Allegretti, and et al. 2022. "Impact of Fecal Microbiota Transplantation on Gut Bacterial Bile Acid Metabolism in Humans" Nutrients 14, no. 24: 5200. https://doi.org/10.3390/nu14245200
APA StyleBustamante, J. -M., Dawson, T., Loeffler, C., Marfori, Z., Marchesi, J. R., Mullish, B. H., Thompson, C. C., Crandall, K. A., Rahnavard, A., Allegretti, J. R., & Cummings, B. P. (2022). Impact of Fecal Microbiota Transplantation on Gut Bacterial Bile Acid Metabolism in Humans. Nutrients, 14(24), 5200. https://doi.org/10.3390/nu14245200