
Selenium Deficiency during Pregnancy in Mice Impairs Exercise Performance and Metabolic Function in Adult Offspring



Abstract
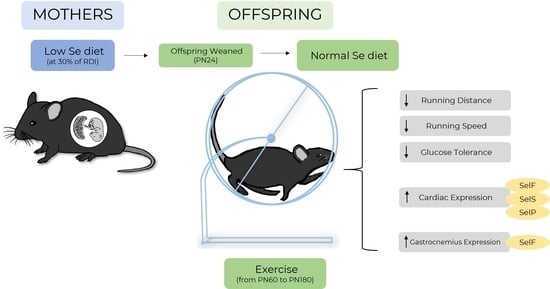
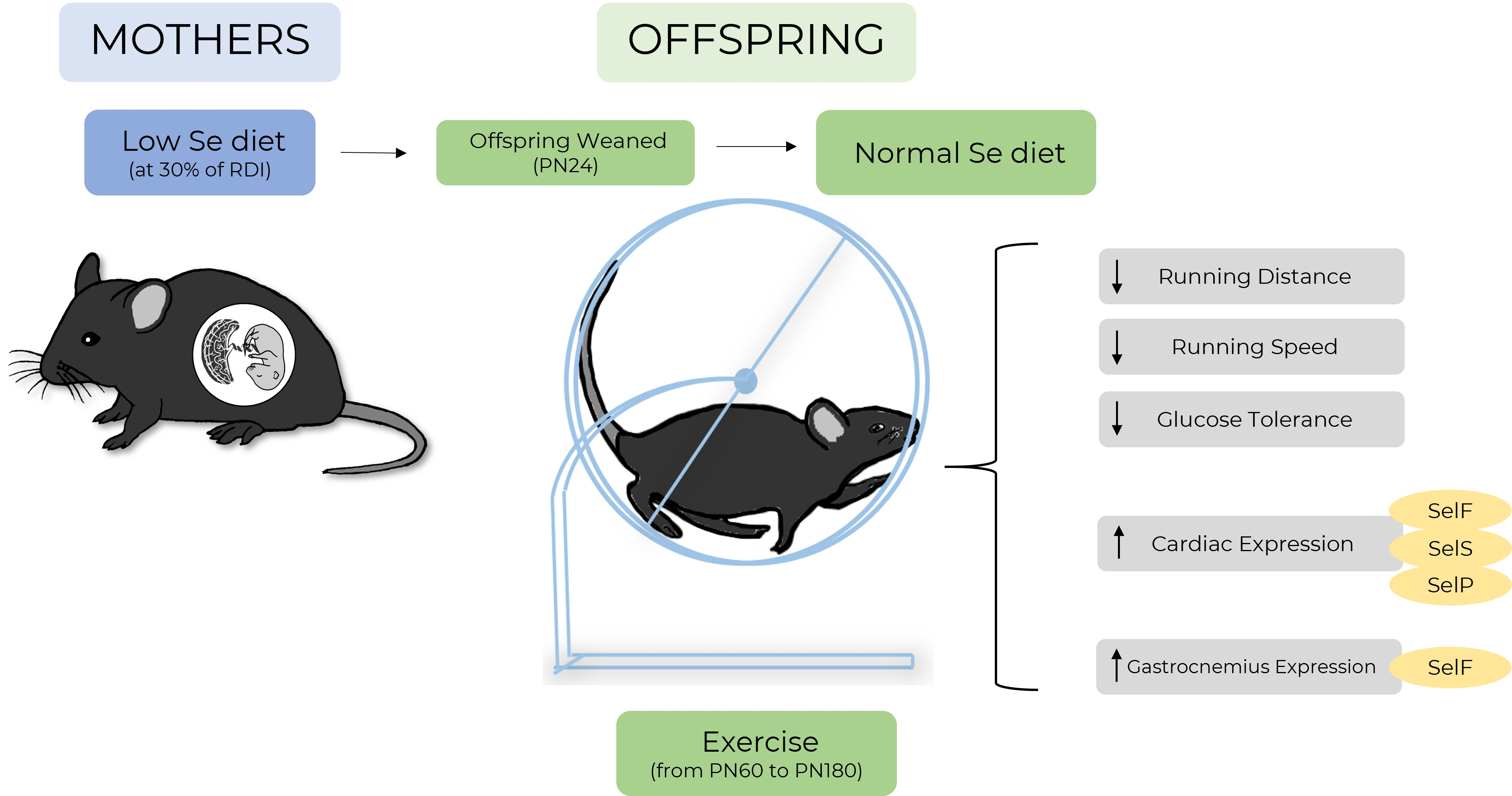
:
1. Introduction
2. Materials and Methods
2.1. Animal Procedures
2.2. Mitochondrial Respiration
2.3. Quantitative PCR
2.4. Statistical Analysis
3. Results
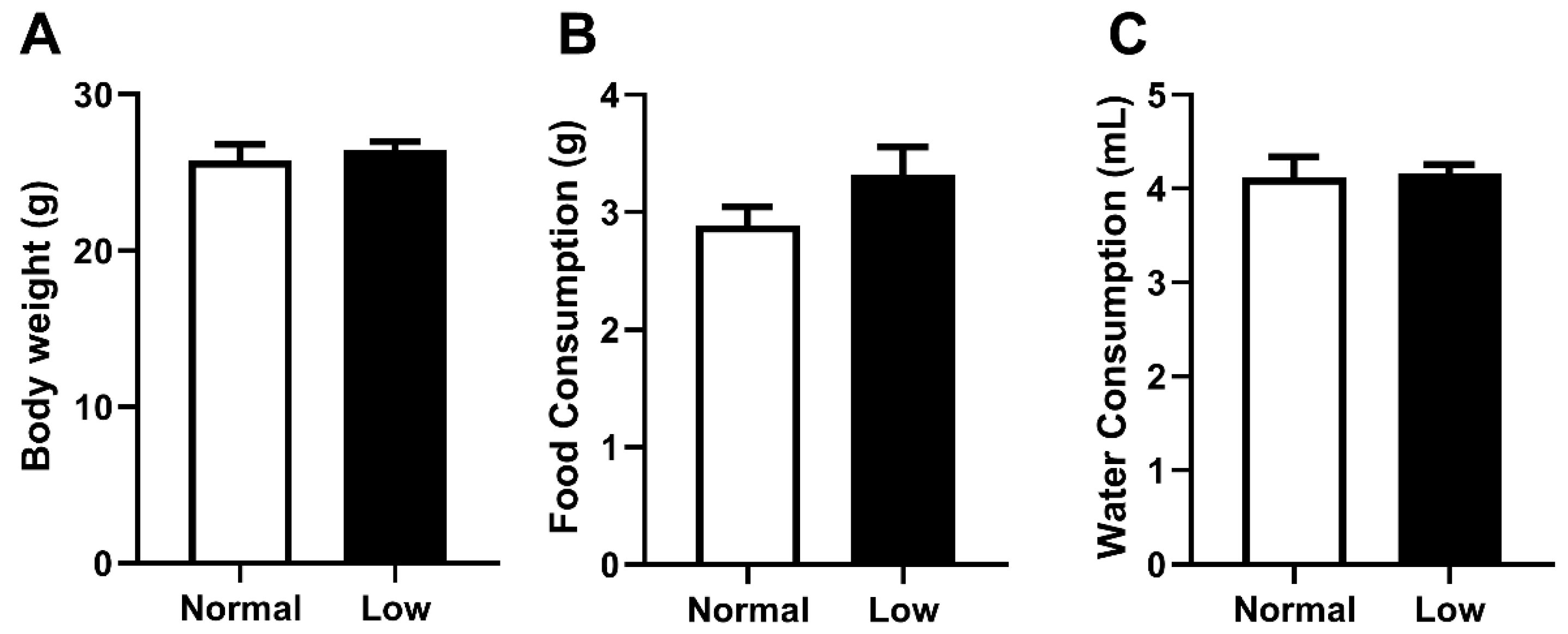
3.1. Offspring Weight, Food and Water Consumption
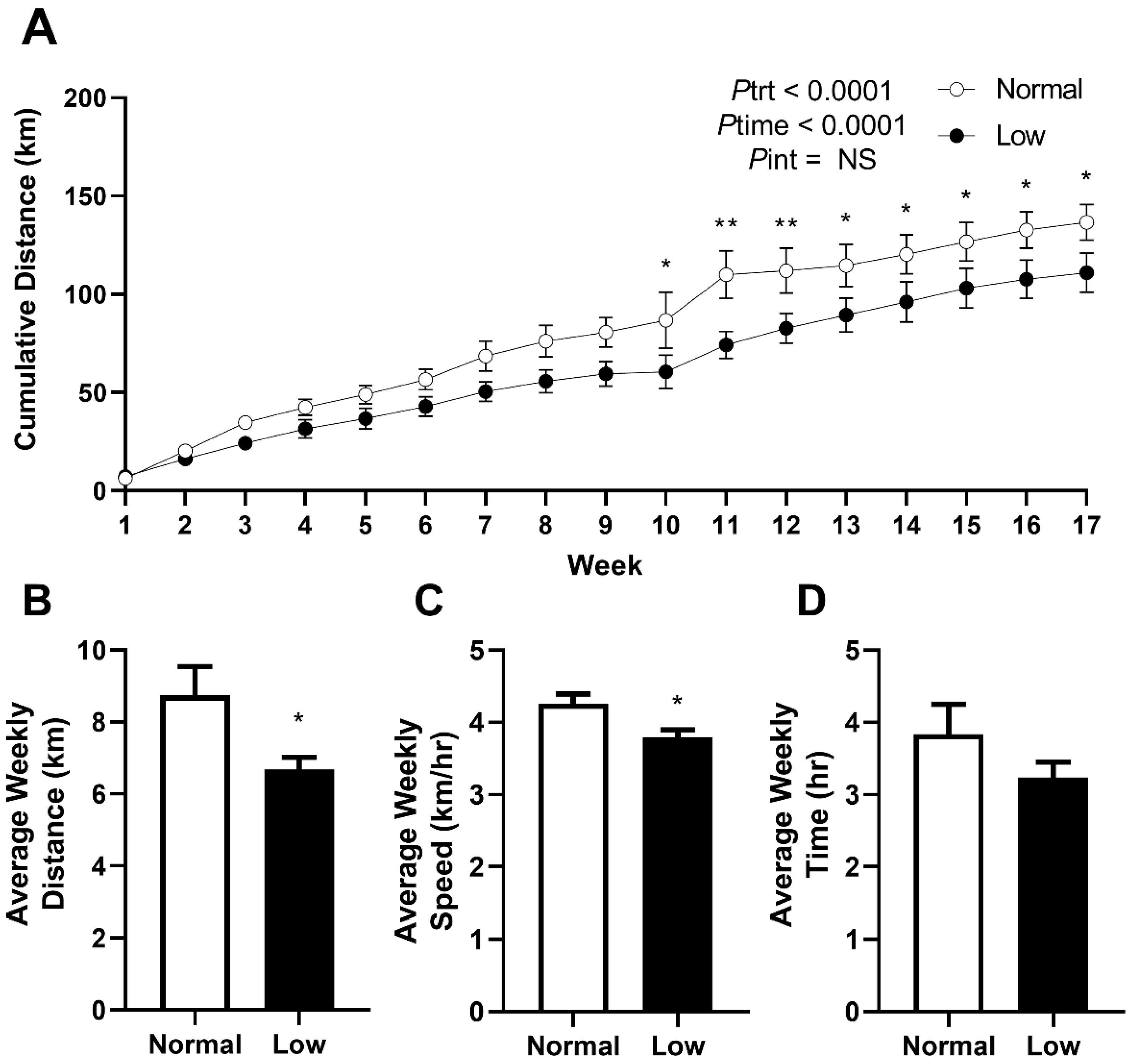
3.2. Exercise Behavior
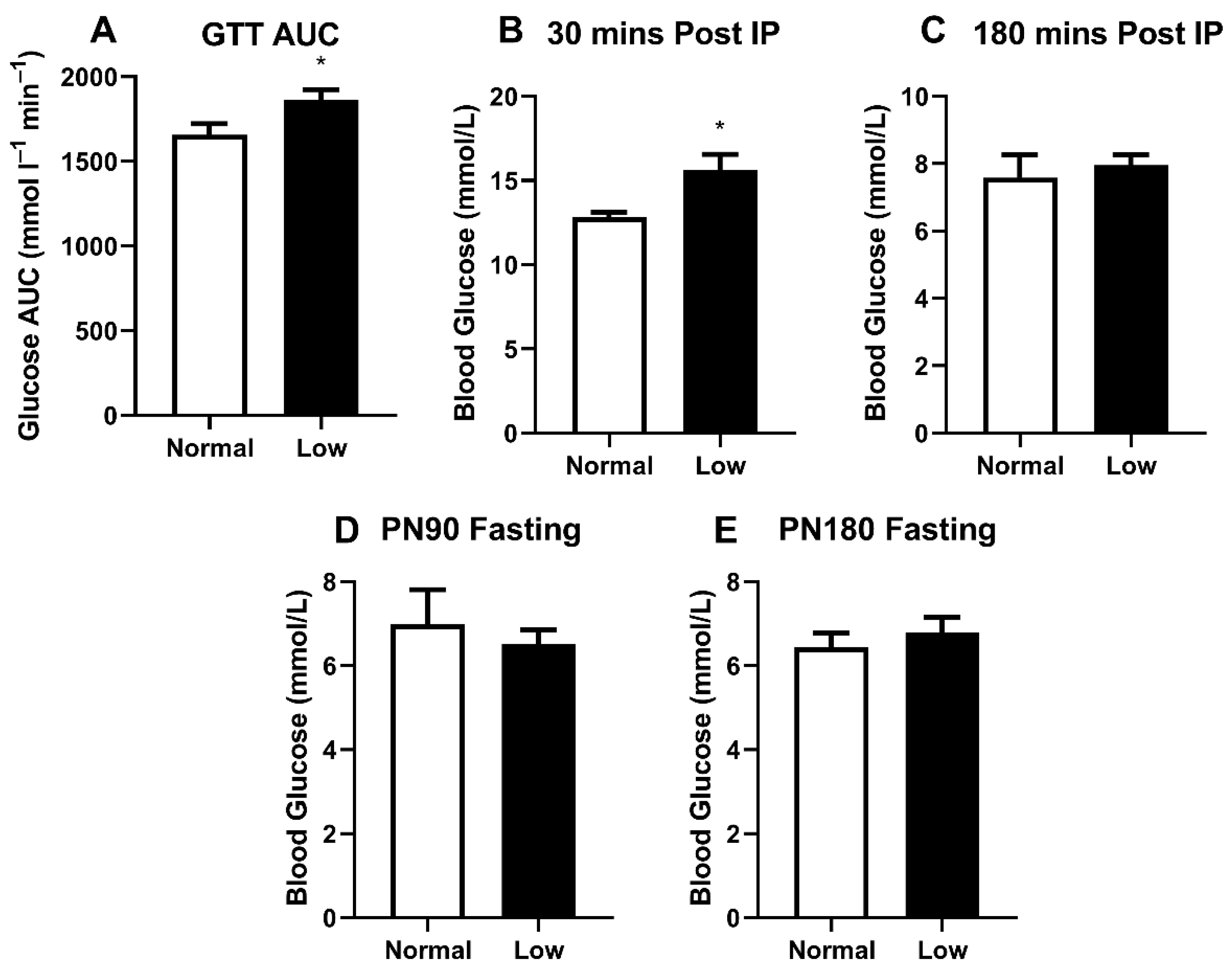
3.3. Glucose Metabolism
3.4. Selenoprotein Expression in the Heart and Gastrocnemii
3.5. Mitochondrial Respiration of the Heart
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Silverman, M.N.; Deuster, P.A. Biological mechanisms underlying the role of physical fitness in health and resilience. Interface Focus 2014, 4, 20140040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, H.N.; Chen, C.C.; Hood, D.A. Mitochondria, muscle health, and exercise with advancing age. Physiology 2015, 30, 208–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heath, G.; Gavin, J., 3rd; Hinderliter, J.; Hagberg, J.; Bloomfield, S.; Holloszy, J. Effects of exercise and lack of exercise on glucose tolerance and insulin sensitivity. J. Appl. Physiol. 1983, 55, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Pitsavos, C.; Panagiotakos, D.; Weinem, M.; Stefanadis, C. Diet, exercise and the metabolic syndrome. Rev. Diabet. Stud. 2006, 3, 118. [Google Scholar] [CrossRef] [Green Version]
- Kern, H.J.; Mitmesser, S.H. Role of nutrients in metabolic syndrome: A 2017 update. Nutr. Diet. Suppl. 2018, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Baltaci, A.K.; Mogulkoc, R.; Akil, M.; Bicer, M. Selenium: Its metabolism and relation to exercise. Pak. J. Pharm. Sci. 2016, 29, 1719–1725. [Google Scholar]
- Hofstee, P.; McKeating, D.; Perkins, A.V.; Cuffe, J.S. Placental adaptations to micronutrient dysregulation in the programming of chronic disease. Clin. Exp. Pharmacol. Physiol. 2018, 45, 871–884. [Google Scholar] [CrossRef]
- Rederstorff, M.; Krol, A.; Lescure, A. Understanding the importance of selenium and selenoproteins in muscle function. Cell. Mol. Life Sci. 2006, 63, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Chen, J. An original discovery: Selenium deficiency and Keshan disease (an endemic heart disease). Asia Pac. J. Clin. Nutr. 2012, 21, 320. [Google Scholar]
- Valberg, S.J. Chapter 10—Disorders of the Musculoskeletal System. In Equine Internal Medicine, 4th ed.; Reed, S.M., Bayly, W.M., Sellon, D.C., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2018; pp. 542–579. [Google Scholar] [CrossRef]
- Moghadaszadeh, B.; Petit, N.; Jaillard, C.; Brockington, M.; Roy, S.Q.; Merlini, L.; Romero, N.; Estournet, B.; Desguerre, I.; Chaigne, D. Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nat. Genet. 2001, 29, 17. [Google Scholar] [CrossRef]
- Addinsall, A.B.; Wright, C.R.; Kotsiakos, T.L.; Smith, Z.M.; Cook, T.R.; Andrikopoulos, S.; van der Poel, C.; Stupka, N. Impaired exercise performance is independent of inflammation and cellular stress following genetic reduction or deletion of selenoprotein S. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R981–R996. [Google Scholar] [CrossRef] [PubMed]
- Mikovic, J.; Lamon, S. The effect of maternal metabolic status on offspring health: A role for skeletal muscle? J. Physiol. 2018, 596, 5079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofstee, P.; McKeating, D.R.; Bartho, L.A.; Anderson, S.T.; Perkins, A.V.; Cuffe, J.S.J.N. Maternal Selenium Deficiency in Mice Alters Offspring Glucose Metabolism and Thyroid Status in a Sexually Dimorphic Manner. Nutrients 2020, 12, 267. [Google Scholar] [CrossRef] [Green Version]
- Hofstee, P.; Cuffe, J.S.; Perkins, A.V. Analysis of Selenoprotein Expression in Response to Dietary Selenium Deficiency during Pregnancy Indicates Tissue Specific Differential Expression in Mothers and Sex Specific Changes in the Fetus and Offspring. Int. J. Mol. Sci. 2020, 21, 2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, M.; Thamotharan, M.; Oak, S.A.; Pan, G.; Maclaren, D.C.; Lee, P.W.; Devaskar, S.U. Early exercise regimen improves insulin sensitivity in the intrauterine growth-restricted adult female rat offspring. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E272–E281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, J.L.; Huber, K.; Thompson, N.M.; Davison, M.; Breier, B.H. Moderate daily exercise activates metabolic flexibility to prevent prenatally induced obesity. Endocrinology 2009, 150, 179–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofstee, P.; Bartho, L.A.; McKeating, D.R.; Radenkovic, F.; McEnroe, G.; Fisher, J.J.; Holland, O.J.; Vanderlelie, J.J.; Perkins, A.V.; Cuffe, J.S. Maternal selenium deficiency during pregnancy in mice increases thyroid hormone concentrations, alters placental function and reduces fetal growth. J. Physiol. 2019, 597, 5587–5617. [Google Scholar] [CrossRef]
- Fasching, M.; Fontana-Ayoub, M.; Gnaiger, E. Mitochondrial respiration medium-MiR06. Mitochondrial Physiol. Netw. 2014, 14, 1–5. [Google Scholar]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments; Oxford University Press: Oxford, UK, 2009; pp. 611–622. [Google Scholar]
- Golbidi, S.; Mesdaghinia, A.; Laher, I. Exercise in the metabolic syndrome. Oxidative Med. Cell. Longev. 2012, 2012, 349710. [Google Scholar] [CrossRef] [Green Version]
- Novak, C.M.; Burghardt, P.R.; Levine, J.A. The use of a running wheel to measure activity in rodents: Relationship to energy balance, general activity, and reward. Neurosci. Biobehav. Rev. 2012, 36, 1001–1014. [Google Scholar] [CrossRef] [Green Version]
- Stapleton, D.I.; Lau, X.; Flores, M.; Trieu, J.; Gehrig, S.M.; Chee, A.; Naim, T.; Lynch, G.S.; Koopman, R. Dysfunctional muscle and liver glycogen metabolism in mdx dystrophic mice. PLoS ONE 2014, 9, e91514. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, H.; Men, L.-l.; Huang, R.-c.; Zhou, H.-c.; Xing, Q.; Yao, J.-j.; Shi, C.-h.; Du, J.-l. Effects of selenoprotein S on oxidative injury in human endothelial cells. J. Transl. Med. 2013, 11, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, B.; Liu, M.; Ni, J.; Tian, J. Role of Selenoprotein F in Protein Folding and Secretion: Potential Involvement in Human Disease. Nutrients 2018, 10, 1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellinger, F.P.; Raman, A.V.; Reeves, M.A.; Berry, M.J. Regulation and function of selenoproteins in human disease. Biochem. J. 2009, 422, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Soumya, R.S.; Prathapan, A.; Raj, P.S.; Vineetha, V.P.; Raghu, K.G. Selenium incorporated guar gum nanoparticles safeguard mitochondrial bioenergetics during ischemia reperfusion injury in H9c2 cardiac cells. Int. J. Biol. Macromol. 2018, 107, 254–260. [Google Scholar] [CrossRef]
- Mehta, S.L.; Kumari, S.; Mendelev, N.; Li, P.A. Selenium preserves mitochondrial function, stimulates mitochondrial biogenesis, and reduces infarct volume after focal cerebral ischemia. BMC Neurosci. 2012, 13, 79. [Google Scholar] [CrossRef] [Green Version]
- Neal, E.S.; Hofstee, P.; Askew, M.R.; Kent, N.L.; Bartho, L.A.; Perkins, A.V.; Cuffe, J.S.M. Maternal selenium deficiency in mice promotes sex-specific changes to urine flow and renal expression of mitochondrial proteins in adult offspring. Physiol. Rep. 2021, 9, e14785. [Google Scholar] [CrossRef]
- Ylli, D.; Klubo-Gwiezdzinska, J.; Wartofsky, L. Exercise and Thyroid Function. In Endocrinology of Physical Activity and Sport; Hackney, A.C., Constantini, N.W., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 85–108. [Google Scholar] [CrossRef]
- Parra-Montes de Oca, M.A.; Gutierrez-Mariscal, M.; Salmeron-Jimenez, M.F.; Jaimes-Hoy, L.; Charli, J.L.; Joseph-Bravo, P. Voluntary Exercise-Induced Activation of Thyroid Axis and Reduction of White Fat Depots Is Attenuated by Chronic Stress in a Sex Dimorphic Pattern in Adult Rats. Front. Endocrinol. (Lausanne) 2019, 10, 418. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Gene Name | Gene Symbol | Accession Number | Primer Sequence |
|---|---|---|---|---|
| Selenoproteins | Thioredoxin Reductase 1 | Txnrd1 | NM_001042513 | F’ TCCCAACGAAAATTGAACAG R’ TGTTAAATTCGCCCTCTATG |
| Thioredoxin Reductase 2 | Txnrd2 | NM_013711 | F’ GAATCACAAGTGACGACATC R’ AAAGATGACATTTGCTGGTC | |
| Glutathione Peroxidase 1 | Gpx1 | NM_008160 | F’ GGAGAATGGCAAGAATGAAG R’ TTCGCACTTCTCAAACAATG | |
| Glutathione Peroxidase 3 | Gpx3 | NM_008161 | F’ ACAAGAGAAGTCTAAGACAGAC R’ TGTAGTGCATTCAGTTCAAG | |
| Iodothyronine Deiodinase Type 2 | Dio2 | NM_010050 | F’ CAGTCTTTTTCTCCAACTGC R’ CCAGTTTAACCTGTTTGTAGG | |
| Selenoprotein F | SelenoF | NM_053102 | F’ CTACAGATCAAGTATGTTCGAG R’ TATATGCGTTCCAACTTCTC | |
| Selenoprotein S | SelenoS | NM_024439 | F’ ACCTGATGTTGTTGTTAGC R’ CTCTTCTTCAAGCTGTCTTAG | |
| Selenoprotein K | SelenoK | NM_019979 | F’ TGATTCCAGATACGACGATG R’ CATTTACCTTCCTCATCCAC | |
| Selenoprotein M | SelenoM | NM_053267 | F’ GACAGTTGAATCGCCTAAAG R’ TGGTAATTTCGGCTTAACAG | |
| Selenoprotein T | SelenoT | NM_001040396 | F’ GTTCCAGATTTGTGTATCCTG R’ GTGTCTATAAATTGGTTGAGGG | |
| Selenoprotein N | SelenoN | NM_029100 | F’ CTTCAAGAAGGTCAACTACC R’ AGCAAGATGGAATGAACAAG | |
| Selenoprotein P | SelenoP | NM_001042613 | F’ ATGACTTCCTCATCTATGACAG R’ GAGGTCACAGTTTACAGAAG | |
| House Keepers | Beta-Actin | Actb | NM_007393 | F’ GATGTATGAAGGCTTTGGTC R’ TGTGCACTTTTATTGGTCTC |
| Beta-2-Microglobulin | B2m | NM_009735 | F’ GTATGCTATCCAGAAAACCC R’ CTGAAGGACATATCTGACATC | |
| Hypoxanthine Phosphoribosyltransferase 1 | Hprt1 | NM_013556 | F’ AGGGATTTGAATCACGTTTG R’ TTTACTGGCAACATCAACAG |
| Normal | Low | p | |
|---|---|---|---|
| Heart | 149.15 ± 0.10 | 186.90 ± 19.4 | NS |
| Liver | 1308.54 ± 48.91 | 1322.95 ± 98.30 | NS |
| Kidney | 160.01 ± 7.11 | 171.63 ± 4.10 | NS |
| Adrenal | 1.64 ± 0.10 | 2.07 ± 0.21 | NS |
| Brain | 419.85 ± 10.46 | 438.82 ± 7.96 | NS |
| Gastrocnemius | 133.06 ± 3.83 | 149.05 ± 8.98 | NS |
| Tibialis Anterior | 42.37 ± 4.00 | 44.98 ± 0.92 | NS |
| Soleus | 9.33 ± 0.73 | 10.17 ± 0.32 | NS |
| EDL | 9.90 ± 0.32 | 9.31 ± 0.66 | NS |
| Testes | 88.97 ± 2.23 | 87.30 ± 3.05 | NS |
| Heart | Gastrocnemius | |||||
|---|---|---|---|---|---|---|
| Normal | Low | p | Normal | Low | p | |
| TrxR1 | 1.00 ± 0.16 | 1.20 ± 0.21 | NS | 1.00 ± 0.43 | 1.12 ± 0.54 | NS |
| TrxR2 | 1.00 ± 0.13 | 1.39 ± 0.25 | NS | 1.00 ± 0.48 | 0.84 ± 0.46 | NS |
| GPx1 | 1.00 ± 0.21 | 1.03 ± 0.17 | NS | 1.00 ± 0.21 | 1.81 ± 0.62 | NS |
| GPx3 | 1.00 ± 0.34 | 0.89 ± 0.32 | NS | 1.00 ± 0.46 | 1.03 ± 0.32 | NS |
| DIO2 | 1.00 ± 0.13 | 0.90 ± 0.24 | NS | - | - | - |
| SelenoF | 1.00 ± 0.24 | 1.83 ± 0.19 | 0.047 | 1.00 ± 0.13 | 1.66 ± 0.26 | 0.030 |
| SelenoS | 1.00 ± 0.19 | 1.95 ± 0.31 | 0.028 | 1.00 ± 0.46 | 1.05 ± 0.25 | NS |
| SelenoK | 1.00 ± 0.25 | 1.09 ± 0.17 | NS | 1.00 ± 0.27 | 1.15 ± 0.35 | NS |
| SelenoM | 1.00 ± 0.27 | 0.87 ± 0.18 | NS | 1.00 ± 0.48 | 1.76 ± 0.82 | NS |
| SelenoT | 1.00 ± 0.26 | 0.58 ± 0.09 | NS | 1.00 ± 0.20 | 0.99 ± 0.18 | NS |
| SelenoN | 1.00 ± 0.37 | 0.93 ± 0.29 | NS | - | - | - |
| SelenoP | 1.00 ± 0.25 | 1.82 ± 0.10 | 0.030 | 1.00 ± 0.18 | 1.42 ± 0.20 | NS |
| Normal | Low | p | ||
|---|---|---|---|---|
| Fluxes | CI | 140.61 ± 56.68 | 88.66 ± 40.43 | NS |
| Cic | 210.22 ± 68.65 | 173.20 ± 67.00 | NS | |
| CII | 523.47 ± 60.75 | 381.87 ± 108.53 | NS | |
| ETS | 517.33 ± 61.40 | 402.62 ± 120.83 | NS | |
| CIV | 832.62 ± 146.89 | 1017.73 ± 242.13 | NS | |
| ROX | 7.18 ± 2.29 | 7.89 ± 4.78 | NS | |
| Flux control factors (FCF) | FCF CI | 0.24 ± 0.08 | 0.20 ± 0.04 | NS |
| FCF CI + CII | 0.78 ± 0.08 | 0.78 ± 0.06 | NS | |
| FCFc | 0.45 ± 0.08 | 0.41 ± 0.10 | NS | |
| L/ETS | 0.04 ± 0.00 | 0.11 ± 0.04 | NS | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hofstee, P.; Perkins, A.V.; Cuffe, J.S.M. Selenium Deficiency during Pregnancy in Mice Impairs Exercise Performance and Metabolic Function in Adult Offspring. Nutrients 2022, 14, 1125. https://doi.org/10.3390/nu14051125
Hofstee P, Perkins AV, Cuffe JSM. Selenium Deficiency during Pregnancy in Mice Impairs Exercise Performance and Metabolic Function in Adult Offspring. Nutrients. 2022; 14(5):1125. https://doi.org/10.3390/nu14051125
Chicago/Turabian StyleHofstee, Pierre, Anthony V. Perkins, and James S. M. Cuffe. 2022. "Selenium Deficiency during Pregnancy in Mice Impairs Exercise Performance and Metabolic Function in Adult Offspring" Nutrients 14, no. 5: 1125. https://doi.org/10.3390/nu14051125
APA StyleHofstee, P., Perkins, A. V., & Cuffe, J. S. M. (2022). Selenium Deficiency during Pregnancy in Mice Impairs Exercise Performance and Metabolic Function in Adult Offspring. Nutrients, 14(5), 1125. https://doi.org/10.3390/nu14051125