
PKN1 Kinase: A Key Player in Adipocyte Differentiation and Glucose Metabolism

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Patient Samples Processing
2.3. Reagents and Antibodies
2.4. Cell Culture, Adipocyte Differentiation and Cell Area Measurements
2.5. Western Blot Analysis
2.6. Oil Red O Staining
2.7. 2-[3H] Deoxyglucose Uptake Assay in 3T3-L1-Differntiated Adipocytes
2.8. Lentivirus Packaging and Infection and Generation of Stably Transduced 3T3L1 Cells
2.9. RNA Isolation and Gene Expression Analysis by RT-PCR
2.10. Statistical Analysis
3. Results
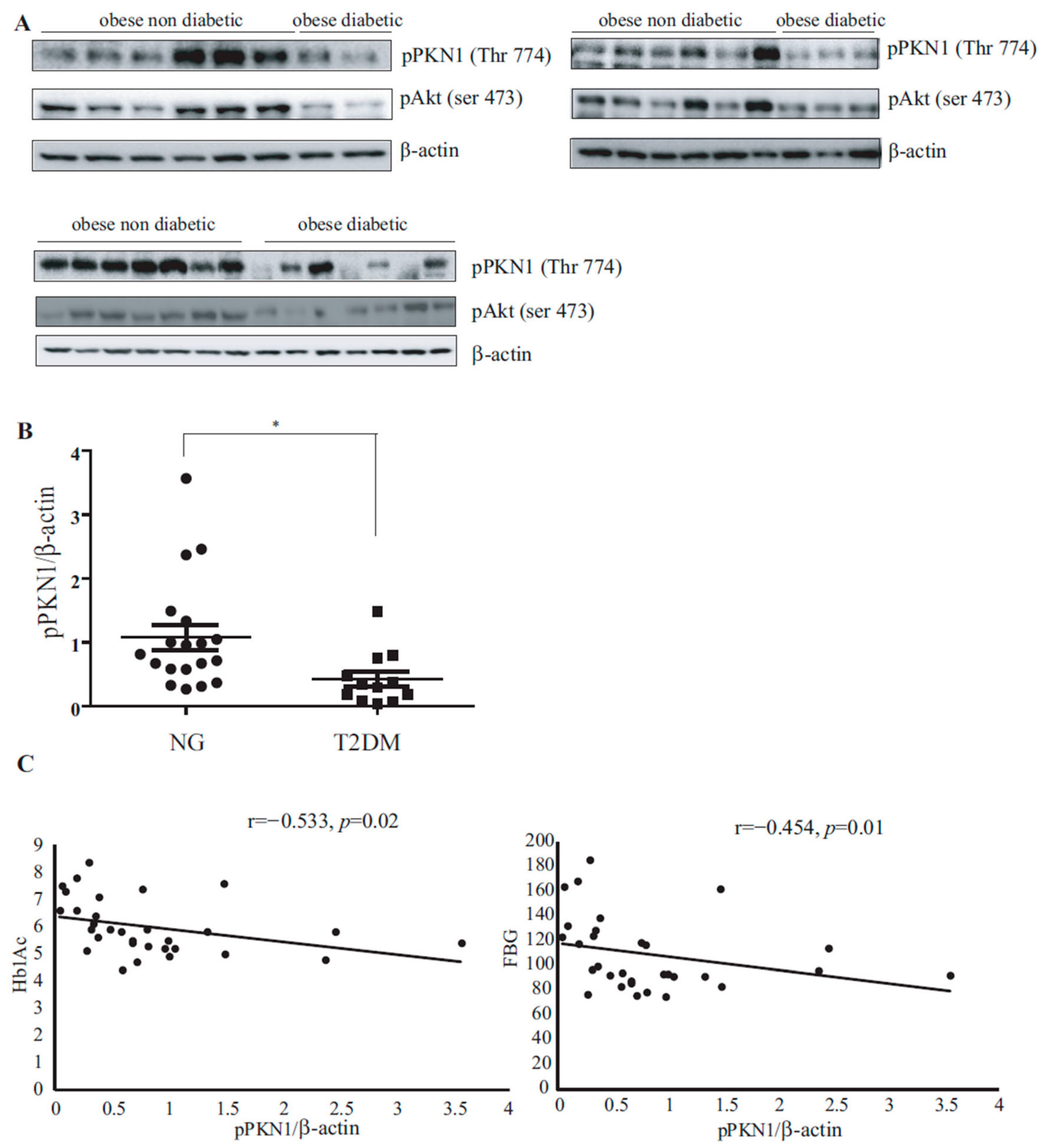
3.1. PKN1 Activation Levels in VAT of Patients with Obesity Inversely Correlate with T2DM Presence
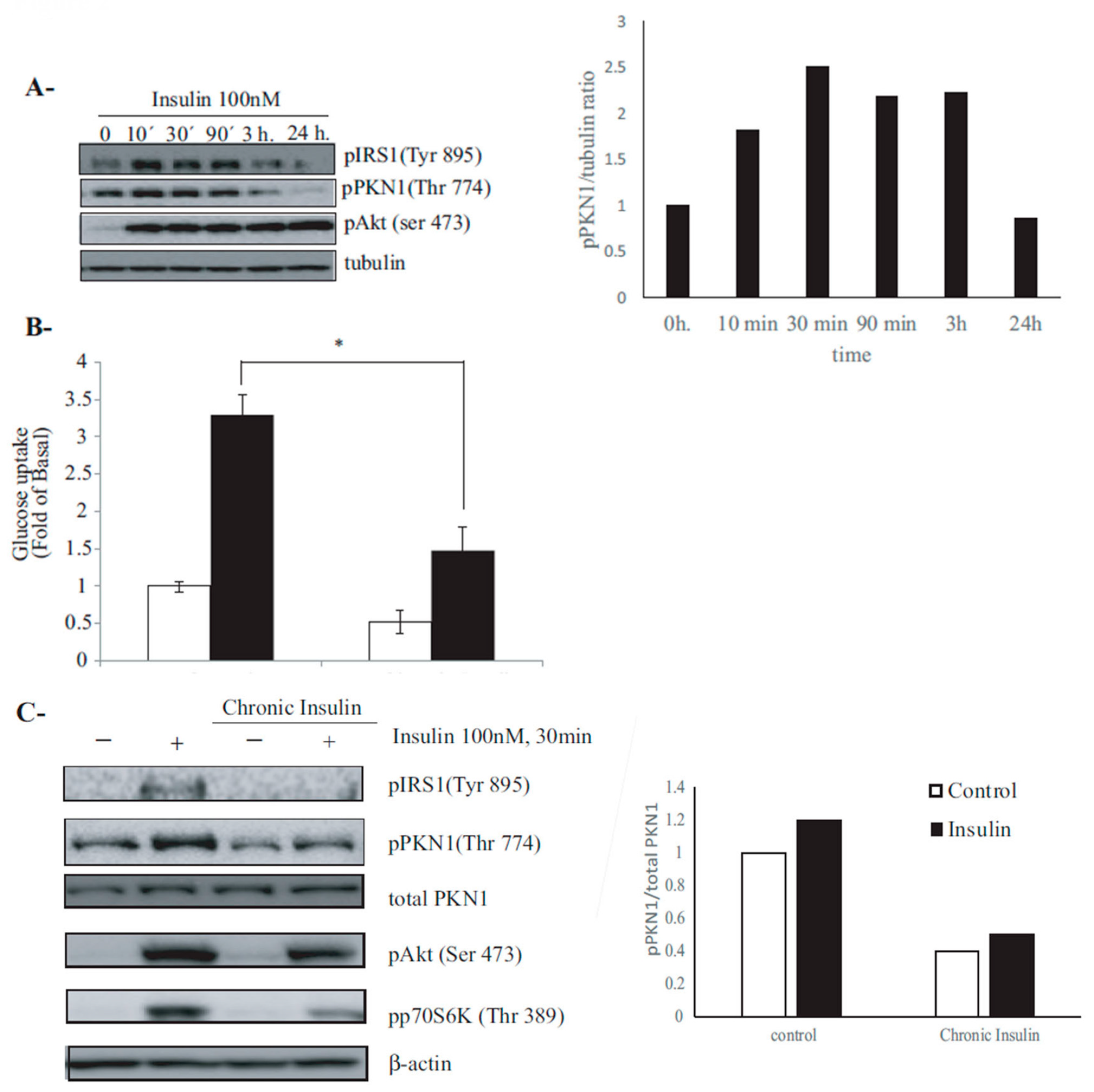
3.2. PKN1 Is Phosphorylated by Insulin Signaling in 3T3-L1 Adipocytes, and This Phosphorylation Is Hindered under Insulin-Resistance Conditions
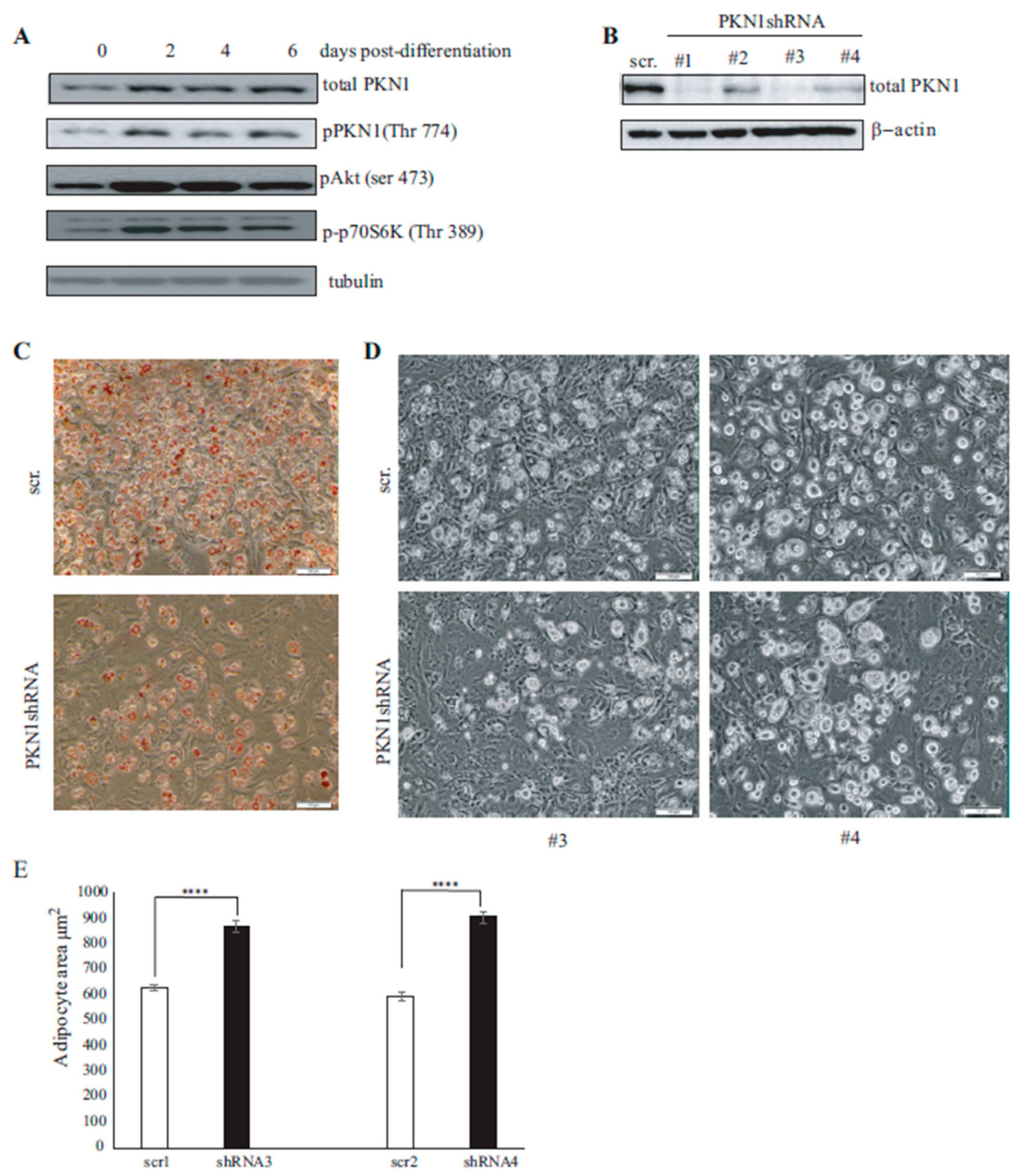
3.3. PKN1 Is Activated during Adipocyte Differentiation and Is Required for Full Adipocyte Differentiation
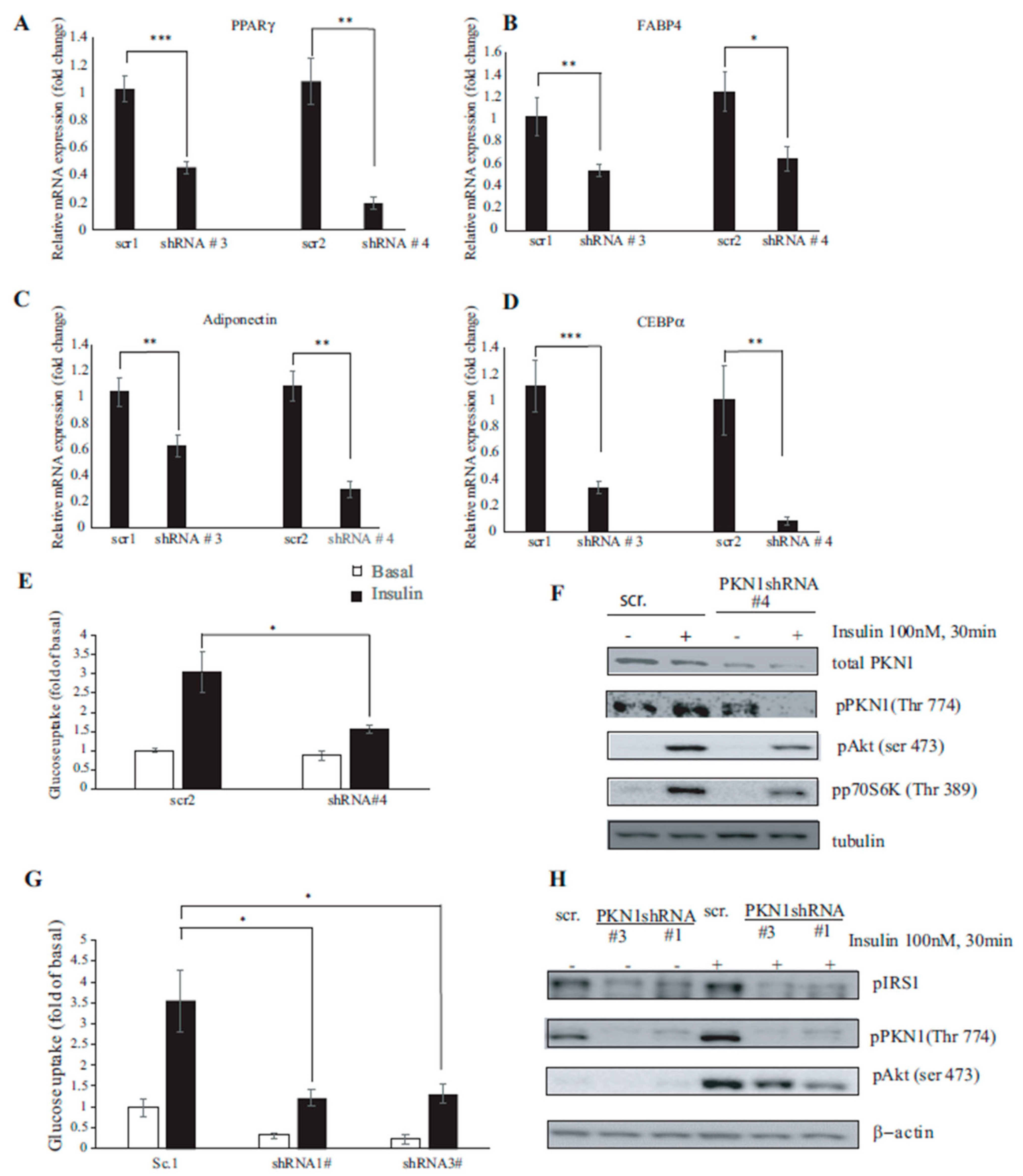
3.4. PKN1 Is Required for Full Adipocyte Differentiation
3.5. PKN1 Knockdown Impairs Insulin Signaling and Glucose Uptake in 3T3-L1 Adipocytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef]
- Van Gaal, L.F.; Mertens, I.L.; De Block, C.E. Mechanisms linking obesity with cardiovascular disease. Nature 2006, 444, 875–880. [Google Scholar] [CrossRef]
- Abel, E.D.; Peroni, O.; Kim, J.K.; Kim, Y.B.; Boss, O.; Hadro, E.; Minnemann, T.; Shulman, G.I.; Kahn, B.B. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 2001, 409, 729–733. [Google Scholar] [CrossRef]
- Standaert, M.L.; Galloway, L.; Karnam, P.; Bandyopadhyay, G.; Moscat, J.; Farese, R.V. Protein kinase C-zeta as a downstream effector of phosphatidylinositol 3-kinase during insulin stimulation in rat adipocytes. Potential role in glucose transport. J. Biol. Chem. 1997, 272, 30075–30082. [Google Scholar] [CrossRef]
- Le Good, J.A.; Ziegler, W.H.; Parekh, D.B.; Alessi, D.R.; Cohen, P.; Parker, P.J. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science 1998, 281, 2042–2045. [Google Scholar] [CrossRef]
- Mellor, H.; Parker, P.J. The extended protein kinase C superfamily. Biochem. J. 1998, 332 (Pt 2), 281–292. [Google Scholar] [CrossRef]
- Dekker, L.V.; Palmer, R.H.; Parker, P.J. The protein kinase C and protein kinase C related gene families. Curr. Opin. Struct. Biol. 1995, 5, 396–402. [Google Scholar] [CrossRef]
- Nolen, B.; Taylor, S.; Ghosh, G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol. Cell 2004, 15, 661–675. [Google Scholar] [CrossRef]
- Flynn, P.; Mellor, H.; Casamassima, A.; Parker, P.J. Rho GTPase control of protein kinase C-related protein kinase activation by 3-phosphoinositide-dependent protein kinase. J. Biol. Chem. 2000, 275, 11064–11070. [Google Scholar] [CrossRef]
- Lachmann, S.; Jevons, A.; De Rycker, M.; Casamassima, A.; Radtke, S.; Collazos, A.; Parker, P.J. Regulatory domain selectivity in the cell-type specific PKN-dependence of cell migration. PLoS ONE 2011, 6, e21732. [Google Scholar] [CrossRef]
- David, M.; Petit, D.; Bertoglio, J. Cell cycle regulation of Rho signaling pathways. Cell Cycle 2012, 11, 3003–3310. [Google Scholar] [CrossRef]
- Standaert, M.; Bandyopadhyay, G.; Galloway, L.; Ono, Y.; Mukai, H.; Farese, R. Comparative effects of GTPgammaS and insulin on the activation of Rho, phosphatidylinositol 3-kinase, and protein kinase N in rat adipocytes. Relationship to glucose transport. J. Biol. Chem. 1998, 273, 7470–7477. [Google Scholar] [CrossRef]
- Dong, L.Q.; Landa, L.R.; Wick, M.J.; Zhu, L.; Mukai, H.; Ono, Y.; Liu, F. Phosphorylation of protein kinase N by phosphoinositide-dependent protein kinase-1 mediates insulin signals to the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 2000, 97, 5089–5094. [Google Scholar] [CrossRef]
- American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2021. Diabetes Care 2021, 44 (Suppl. S1), S15–S33. [Google Scholar] [CrossRef]
- Shibata, M.; Hakuno, F.; Yamanaka, D.; Okajima, H.; Fukushima, T.; Hasegawa, T.; Ogata, T.; Toyoshima, Y.; Chida, K.; Kimura, K.; et al. Paraquat-induced oxidative stress represses phosphatidylinositol 3-kinase activities leading to impaired glucose uptake in 3T3-L1 adipocytes. J. Biol. Chem. 2010, 285, 20915–20925. [Google Scholar] [CrossRef]
- Hoehn, K.L.; Hohnen-Behrens, C.; Cederberg, A.; Wu, L.E.; Turner, N.; Yuasa, T.; Yousuke, E.; David, E.J. IRS1-independent defects define major nodes of insulin resistance. Cell Metab. 2008, 7, 421–433. [Google Scholar] [CrossRef]
- Lopez-Mejia, I.C.; Pijuan, J.; Navaridas, R.; Santacana, M.; Gatius, S.; Velasco, A.; Castellà, G.; Panosa, A.; Cabiscol, E.; Pinyol, M.; et al. Oxidative stress-induced FAK activation contributes to uterine serous carcinoma aggressiveness. Mol. Oncol. 2022, 17, 98–118. [Google Scholar] [CrossRef]
- Zebisch, K.; Voigt, V.; Wabitsch, M.; Brandsch, M. Protocol for effective differentiation of 3T3-L1 cells to adipocytes. Anal. Biochem. 2012, 425, 88–90. [Google Scholar] [CrossRef]
- Huang, P.; Altshuller, Y.M.; Hou, J.C.; Pessin, J.E.; Frohman, M.A. Insulin-stimulated plasma membrane fusion of Glut4 glucose transporter-containing vesicles is regulated by phospholipase D1. Mol. Biol. Cell 2005, 16, 2614–2623. [Google Scholar] [CrossRef]
- Yeramian, A.; Santacana, M.; Sorolla, A.; Llobet, D.; Encinas, M.; Velasco, A.; Bahi, N.; Eritja, N.; Domingo, M.; Oliva, E.; et al. Nuclear factor-κB2/p100 promotes endometrial carcinoma cell survival under hypoxia in a HIF-1α independent manner. Lab. Investig. 2011, 91, 859–871. [Google Scholar] [CrossRef]
- Yeramian, A.; Vea, A.; Benítez, S.; Ribera, J.; Domingo, M.; Santacana, M.; Martinez, M.; Maiques, O.; Valls, J.; Dolcet, X.; et al. 2-phenylethynesulphonamide (PFT-μ) enhances the anticancer effect of the novel hsp90 inhibitor NVP-AUY922 in melanoma, by reducing GSH levels. Pigment. Cell Melanoma Res. 2016, 29, 352–371. [Google Scholar] [CrossRef]
- Mlinar, B.; Marc, J. New insights into adipose tissue dysfunction in insulin resistance. Clin. Chem. Lab. Med. 2011, 49, 1925–1935. [Google Scholar] [CrossRef]
- Rosen, E.D.; MacDougald, O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006, 7, 885–896. [Google Scholar] [CrossRef]
- Majithia, A.R.; Flannick, J.; Shahinian, P.; Guo, M.; Bray, M.A.; Fontanillas, P.; Gabriel, S.B.; Rosen, E.D.; Altshuler, D.; Consortium, G.D.; et al. Rare variants in PPARG with decreased activity in adipocyte differentiation are associated with increased risk of type 2 diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 13127–13132. [Google Scholar] [CrossRef]
- Wu, Z.; Rosen, E.D.; Brun, R.; Hauser, S.; Adelmant, G.; Troy, A.E.; McKeon, C.; Darlington, G.J.; Spiegelman, B.M. Cross-regulation of C/EBP alpha and PPAR gamma controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol. Cell 1999, 3, 151–158. [Google Scholar] [CrossRef]
- Löffler, M.C.; Mayer, A.E.; Trujillo Viera, J.; Loza Valdes, A.; El-Merahbi, R.; Ade, C.P.; Till, K.; Werner, S.; Anja, S.; Manuela, E. Protein kinase D1 deletion in adipocytes enhances energy dissipation and protects against adiposity. EMBO J. 2018, 37, e99182. [Google Scholar] [CrossRef]
- Chan, D.C.; Watts, G.F.; Sussekov, A.V.; Barrett, P.H.; Yang, Z.; Hua, J.; Song, S. Adipose tissue compartments and insulin resistance in overweight-obese Caucasian men. Diabetes Res. Clin. Pract. 2004, 63, 77–85. [Google Scholar] [CrossRef]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Zhou, Q.L.; Coleman, K.A.; Chouinard, M.; Boese, Q.; Czech, M.P. Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing. Proc. Natl. Acad. Sci. USA 2003, 100, 7569–7574. [Google Scholar] [CrossRef]
- Senoo, H.; Murata, D.; Wai, M.; Arai, K.; Iwata, W.; Sesaki, H.; Iijima, M. KARATE: PKA-induced KRAS4B-RHOA-mTORC2 supercomplex phosphorylates AKT in insulin signaling and glucose homeostasis. Mol. Cell 2021, 81, 4622–4634.e8. [Google Scholar] [CrossRef]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Sano, H.; Kane, S.; Sano, E.; Mîinea, C.P.; Asara, J.M.; Lane, W.S.; Garner, C.W.; Lienhard, G.E. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J. Biol. Chem. 2003, 278, 14599–14602. [Google Scholar] [CrossRef]
- Oishi, K.; Takahashi, M.; Mukai, H.; Banno, Y.; Nakashima, S.; Kanaho, Y.; Nozawa, Y.; Ono, Y. PKN regulates phospholipase D1 through direct interaction. J. Biol. Chem. 2001, 276, 18096–18101. [Google Scholar] [CrossRef]
- Kitagawa, M.; Shibata, H.; Toshimori, M.; Mukai, H.; Ono, Y. The role of the unique motifs in the amino-terminal region of PKN on its enzymatic activity. Biochem. Biophys. Res. Commun. 1996, 220, 963–968. [Google Scholar] [CrossRef]
- Yang, C.S.; Melhuish, T.A.; Spencer, A.; Ni, L.; Hao, Y.; Jividen, K.; Harris, T.E.; Snow, C.; Frierson, H.F.; Wotton, D.; et al. The protein kinase C super-family member PKN is regulated by mTOR and influences differentiation during prostate cancer progression. Prostate 2017, 77, 1452–1467. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All | Type 2 Diabetes | Non-Type 2 Diabetes | p | |
|---|---|---|---|---|
| n | 31 | 12 | 19 | |
| Age (years) | 44.6 ± 11.1 | 50.2 ± 9.9 | 40.6 ± 10.4 | 0.015 |
| BMI (kg/m2) | 43.6 ± 5.7 | 42.4 ± 5.1 | 44.5 ± 6.0 | 0.323 |
| FPG (mmol/L) | 110.6 ± 28.8 | 137.6 ± 23.9 | 91.1 ± 10.2 | <0.001 |
| Hb1Ac (%) | 6.0 ± 1.0 | 7.0 ± 0.8 | 5.3 ± 0.3 | <0.001 |
| Tryglicerides (mg/dL) | 133.2 ± 54.0 | 151.3 ± 67.92 | 123.8 ± 43.9 | 0.175 |
| LDL-cholesterol (mg/dL) | 167.2 ± 30.0 | 108.5 ± 30.7 | 106.4 ± 30.4 | 0.860 |
| HDL-cholesterol (mg/dL) | 46.6 ± 11.3 | 42.0 ± 8.9 | 49.7 ± 11.9 | 0.067 |
| Total cholesterol (mg/dL) | 178.1 ± 33.3 | 177.9 ± 32.1 | 178.3 ± 35.1 | 0.973 |
| pPKN1/β-actin ratio | 0.82 ± 0.14 | 0.42 ± 0.12 | 1.08 ± 0.19 | 0.017 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrerías-González, F.; Yeramian, A.; Baena-Fustegueras, J.A.; Bueno, M.; Fleitas, C.; de la Fuente, M.; Serrano, J.C.E.; Granado-Serrano, A.; Santamaría, M.; Yeramian, N.; et al. PKN1 Kinase: A Key Player in Adipocyte Differentiation and Glucose Metabolism. Nutrients 2023, 15, 2414. https://doi.org/10.3390/nu15102414
Herrerías-González F, Yeramian A, Baena-Fustegueras JA, Bueno M, Fleitas C, de la Fuente M, Serrano JCE, Granado-Serrano A, Santamaría M, Yeramian N, et al. PKN1 Kinase: A Key Player in Adipocyte Differentiation and Glucose Metabolism. Nutrients. 2023; 15(10):2414. https://doi.org/10.3390/nu15102414
Chicago/Turabian StyleHerrerías-González, Fernando, Andrée Yeramian, Juan Antonio Baena-Fustegueras, Marta Bueno, Catherine Fleitas, Maricruz de la Fuente, José C. E. Serrano, Ana Granado-Serrano, Maite Santamaría, Nadine Yeramian, and et al. 2023. "PKN1 Kinase: A Key Player in Adipocyte Differentiation and Glucose Metabolism" Nutrients 15, no. 10: 2414. https://doi.org/10.3390/nu15102414
APA StyleHerrerías-González, F., Yeramian, A., Baena-Fustegueras, J. A., Bueno, M., Fleitas, C., de la Fuente, M., Serrano, J. C. E., Granado-Serrano, A., Santamaría, M., Yeramian, N., Zorzano-Martínez, M., Mora, C., & Lecube, A. (2023). PKN1 Kinase: A Key Player in Adipocyte Differentiation and Glucose Metabolism. Nutrients, 15(10), 2414. https://doi.org/10.3390/nu15102414