

Non-Alcoholic Fatty Liver Disease and Vitamin D in the UK Biobank: A Two-Sample Bidirectional Mendelian Randomisation Study

 , , , , and
, , , , and
Abstract
:1. Introduction
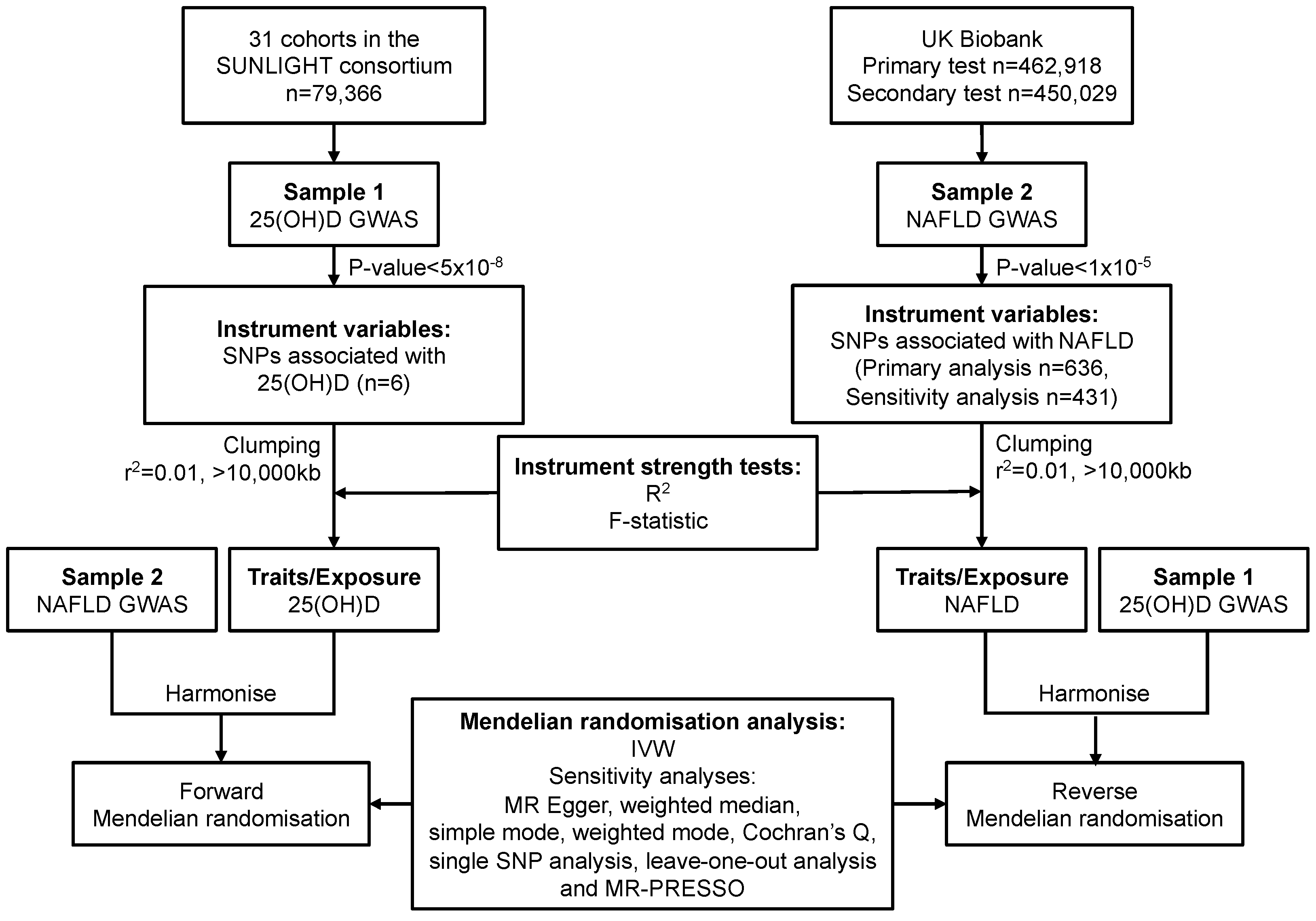
2. Materials and Methods
2.1. Study Design and Data Sources
2.1.1. Data Sources and SNP Selection for Serum 25(OH)D
2.1.2. Data Sources and SNP Selection for NAFLD
2.2. Statistical Power
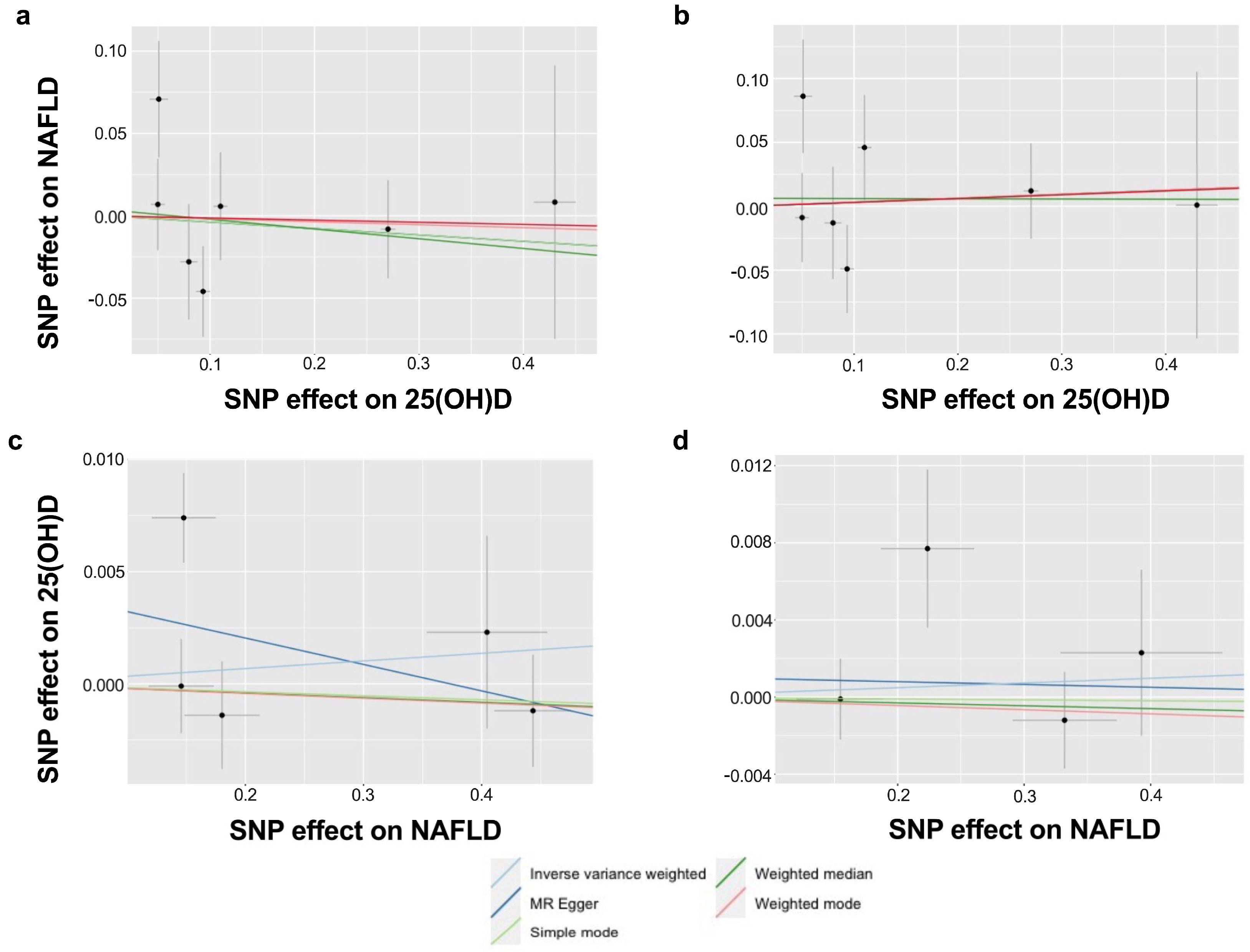
2.3. Mendelian Randomisation Analysis
2.4. Sensitivity Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.M.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Bence, K.K.; Birnbaum, M.J. Metabolic drivers of non-alcoholic fatty liver disease. Mol. Metab. 2021, 50, 101143. [Google Scholar] [CrossRef] [PubMed]
- Zelber-Sagi, S. Dietary Treatment for NAFLD: New Clinical and Epidemiological Evidence and Updated Recommendations. Semin. Liver Dis. 2021, 41, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.B. From sugar to liver fat and public health: Systems biology driven studies in understanding non-alcoholic fatty liver disease pathogenesis. Proc. Nutr. Soc. 2019, 78, 290–304. [Google Scholar] [CrossRef] [Green Version]
- Gibson, P.S.; Quaglia, A.; Dhawan, A.; Wu, H.; Lanham-New, S.; Hart, K.H.; Fitzpatrick, E.; Moore, J.B. Vitamin D status and associated genetic polymorphisms in a cohort of UK children with non-alcoholic fatty liver disease. Pediatr. Obes. 2018, 13, 433–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barchetta, I.; Cimini, F.A.; Cavallo, M.G. Vitamin D and Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD): An Update. Nutrients 2020, 12, 3302. [Google Scholar] [CrossRef]
- Pacifico, L.; Osborn, J.F.; Bonci, E.; Pierimarchi, P.; Chiesa, C. Association between Vitamin D Levels and Nonalcoholic Fatty Liver Disease: Potential Confounding Variables. Mini Rev. Med. Chem. 2019, 19, 310–332. [Google Scholar] [CrossRef]
- Karatayli, E.; Stokes, C.; Lammert, F. Vitamin D in Preclinical Models of Fatty Liver Disease. Anticancer Res. 2020, 40, 527–534. [Google Scholar] [CrossRef]
- Roth, C.L.; Elfers, C.T.; Figlewicz, D.P.; Melhorn, S.J.; Morton, G.J.; Hoofnagle, A.; Yeh, M.M.; Nelson, J.E.; Kowdley, K.V. Vitamin D deficiency in obese rats exacerbates nonalcoholic fatty liver disease and increases hepatic resistin and Toll-like receptor activation. Hepatology 2012, 55, 1103–1111. [Google Scholar] [CrossRef]
- Ma, M.; Long, Q.; Chen, F.; Zhang, T.; Wang, W. Active vitamin D impedes the progression of non-alcoholic fatty liver disease by inhibiting cell senescence in a rat model. Clin. Res. Hepatol. Gastroenterol. 2020, 44, 513–523. [Google Scholar] [CrossRef]
- Rhee, E.-J.; Kim, M.K.; Park, S.E.; Park, C.-Y.; Baek, K.H.; Lee, W.-Y.; Kang, M.I.; Park, S.-W.; Kim, S.-W.; Oh, K.W. High serum vitamin D levels reduce the risk for nonalcoholic fatty liver disease in healthy men independent of metabolic syndrome. Endocr. J. 2013, 60, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Xu, L.; Chen, F.-H.; Zhou, Y.-B. Association of serum vitamin D level and nonalcoholic fatty liver disease: A meta-analysis. Eur. J. Gastroenterol. Hepatol. 2020, 32, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Bertolini, L.; Scala, L.; Cigolini, M.; Zenari, L.; Falezza, G.; Arcaro, G. Associations between serum 25-hydroxyvitamin D3 concentrations and liver histology in patients with non-alcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Eraslan, S.; Kizilgul, M.; Uzunlulu, M.; Colak, Y.; Ozturk, O.; Tuncer, I. Frequency of metabolic syndrome and 25-hydroxyvitamin D3 levels in patients with non-alcoholic fatty liver disease. Minerva Med. 2013, 104, 447–453. [Google Scholar] [PubMed]
- Nelson, J.E.; Roth, C.L.; Wilson, L.; Yates, K.; Aouizerat, B.; Morgan–Stevenson, V.; Whalen, E.; Hoofnagle, A.; Mason, M.; Gersuk, V.; et al. Vitamin D Deficiency Is Associated With Increased Risk of Non-alcoholic Steatohepatitis in Adults with Non-alcoholic Fatty Liver Disease: Possible Role for MAPK and NF-κB? Am. J. Gastroenterol. 2016, 111, 852–863. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Thorne, J.; Moore, J.B. Vitamin D and nonalcoholic fatty liver disease. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 449–458. [Google Scholar] [CrossRef]
- Smith, G.D.; Ebrahim, S. ’Mendelian randomization‘: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 2003, 32, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Sekula, P.; Fabiola Del Greco, M.; Pattaro, C.; Köttgen, A. Mendelian randomization as an approach to assess causality using observational data. J. Am. Soc. Nephrol. 2016, 27, 3253–3265. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Chen, C.; Zhao, L.; Chen, Y.; Han, B.; Xia, F.; Cheng, J.; Li, Q.; Lu, Y. Vitamin D and nonalcoholic fatty liver disease: Bi-directional mendelian randomization analysis. eBioMedicine 2018, 28, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Larsson, S.C. Inverse association between serum 25-hydroxyvitamin D and nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2022, 21, 398–405.e4. [Google Scholar] [CrossRef]
- Jiang, X.; O’Reilly, P.F.; Aschard, H.; Hsu, Y.-H.; Richards, J.B.; Dupuis, J.; Ingelsson, E.; Karasik, D.; Pilz, S.; Berry, D.; et al. Genome-wide association study in 79,366 European-ancestry individuals informs the genetic architecture of 25-hydroxyvitamin D levels. Nat. Commun. 2018, 9, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speliotes, E.K.; Yerges-Armstrong, L.M.; Wu, J.; Hernaez, R.; Kim, L.J.; Palmer, C.D.; Gudnason, V.; Eiriksdottir, G.; Garcia, M.E.; Launer, L.J.; et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011, 7, e1001324. [Google Scholar] [CrossRef]
- 25-Hydroxyvitamin D GWAS Results of the SUNLIGHT Consortium. Available online: https://drive.google.com/drive/folders/0BzYDtCo_doHJRFRKR0ltZHZWZjQ (accessed on 3 March 2022).
- Michaëlsson, K.; Wolk, A.; Byberg, L.; Mitchell, A.; Mallmin, H.; Melhus, H. The seasonal importance of serum 25-hydroxyvitamin D for bone mineral density in older women. J. Intern. Med. 2017, 281, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Sudlow, C.; Gallacher, J.; Allen, N.; Beral, V.; Burton, P.; Danesh, J.; Downey, P.; Elliott, P.; Green, J.; Landray, M.; et al. UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015, 12, e1001779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eslam, M.; George, J. Genetic contributions to NAFLD: Leveraging shared genetics to uncover systems biology. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 40–52. [Google Scholar] [CrossRef]
- Loh, P.-R.; Tucker, G.; Bulik-Sullivan, B.K.; Vilhjálmsson, B.J.; Finucane, H.K.; Salem, R.M.; Chasman, D.I.; Ridker, P.M.; Neale, B.M.; Berger, B.; et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet. 2015, 47, 284–290. [Google Scholar] [CrossRef]
- Loh, P.-R.; Kichaev, G.; Gazal, S.; Schoech, A.P.; Price, A.L. Mixed-model association for biobank-scale datasets. Nat. Genet. 2018, 50, 906–908. [Google Scholar] [CrossRef]
- Howrigan, D. Details and Considerations of the UK BioBank GWAS. Available online: http://www.nealelab.is/blog/2017/9/11/details-and-considerations-of-the-uk-biobank-gwas (accessed on 10 October 2022).
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 2018, 7, e34408. [Google Scholar] [CrossRef]
- R.C. Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 3 March 2022).
- Pritchard, J.K.; Przeworski, M. Linkage disequilibrium in humans: Models and data. Am. J. Hum. Genet. 2001, 69, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhao, Y.; Zhang, H.; Hua, W.; Jiao, W.; Du, X.; Rui, J.; Li, S.; Teng, H.; Shi, B.; et al. Contribution of Rs780094 and Rs1260326 Polymorphisms in GCKR Gene to Non-alcoholic Fatty Liver Disease: A Meta-Analysis Involving 26,552 Participants. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 1696–1708. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.L.; Ahsan, H.; Vander Weele, T.J. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int. J. Epidemiol. 2011, 40, 740–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, S.; Thompson, S.G. Avoiding bias from weak instruments in Mendelian randomization studies. Int.J. Epidemiol. 2011, 40, 755–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef] [Green Version]
- Bowden, J.; Del Greco, M.F.; Minelli, C.; Davey Smith, G.; Sheehan, N.; Thompson, J. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat. Med. 2017, 36, 1783–1802. [Google Scholar] [CrossRef] [Green Version]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef] [Green Version]
- Bowden, J.; Smith, G.D.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, F.P.; Smith, G.D.; Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 2017, 46, 1985–1998. [Google Scholar] [CrossRef] [Green Version]
- Hemani, G.; Bowden, J.; Davey Smith, G. Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum. Mol. Genet. 2018, 27, R195–R208. [Google Scholar] [CrossRef] [Green Version]
- Walker, V.M.; Davies, N.M.; Hemani, G.; Zheng, J.; Haycock, P.C.; Gaunt, T.R.; Davey Smith, G.; Martin, R.M. Using the MR-Base platform to investigate risk factors and drug targets for thousands of phenotypes. Wellcome Open Res. 2019, 4, 113. [Google Scholar] [CrossRef]
- Burgess, S.; Thompson, S.G. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur. J. Epidemiol. 2017, 32, 377–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.F.; Chalumeau, M.; Cohen, R.; Korevaar, D.A.; Khoshnood, B.; Bossuyt, P.M. Cochran’s Q test was useful to assess heterogeneity in likelihood ratios in studies of diagnostic accuracy. J. Clin. Epidemiol. 2015, 68, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Verbanck, M.; Chen, C.-Y.; Neale, B.; Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef]
- Rezaei, S.; Tabrizi, R.; Nowrouzi-Sohrabi, P.; Jalali, M.; Shabani-Borujeni, M.; Modaresi, S.; Gholamalizadeh, M.; Doaei, S. The Effects of Vitamin D Supplementation on Anthropometric and Biochemical Indices in Patients With Non-alcoholic Fatty Liver Disease: A Systematic Review and Meta-analysis. Front. Pharmacol. 2021, 12, 732496. [Google Scholar] [CrossRef]
- Bjelakovic, M.; Nikolova, D.; Bjelakovic, G.; Gluud, C. Vitamin D supplementation for chronic liver diseases in adults. Cochrane Database Syst. Rev. 2021, 8, CD011564. [Google Scholar] [PubMed]
- Zhang, Z. Vitamin D and the Molecular Pathogenesis of Non-alcoholic Fatty Liver Disease. Ph.D. Thesis, University of Leeds, Leeds, UK, 2022. [Google Scholar]
- Hayward, K.L.; Johnson, A.L.; Horsfall, L.U.; Moser, C.; Valery, P.C.; Powell, E.E. Detecting non-alcoholic fatty liver disease and risk factors in health databases: Accuracy and limitations of the ICD-10-AM. BMJ Open Gastroenterol. 2021, 8, e000572. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Darlay, R.; Cockell, S.; Meroni, M.; Govaere, O.; Tiniakos, D.; Burt, A.D.; Bedossa, P.; Palmer, J.; Liu, Y.L.; et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort. J. Hepatol. 2020, 73, 505–515. [Google Scholar] [CrossRef]
- GWAS Results of UK Biobank-Neale Lab. Available online: http://www.nealelab.is/uk-biobank (accessed on 10 October 2022).
- FinnGen R5. Results of Genome-Wide Association Analyses in FinnGen Consortium. 2021. Available online: https://finngen.gitbook.io/documentation/ (accessed on 22 October 2022).
- Bikle, D.D. Vitamin D metabolism, mechanism of action, and clinical applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Yu, K.; Stolzenberg-Solomon, R.; Simon, K.C.; McCullough, M.L.; Gallicchio, L.; Jacobs, E.J.; Ascherio, A.; Helzlsouer, K.; Jacobs, K.B.; et al. Genome-wide association study of circulating vitamin D levels. Hum. Mol. Genet. 2010, 19, 2739–2745. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.J.; Zhang, F.; Richards, J.B.; Kestenbaum, B.; van Meurs, J.B.; Berry, D.; Kiel, D.P.; Streeten, E.A.; Ohlsson, C.; Koller, D.L.; et al. Common genetic determinants of vitamin D insufficiency: A genome-wide association study. Lancet 2010, 376, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, F.P.; Tilling, K.; Smith, G.D.; Lawlor, D.A.; Borges, M.C. Bias in two-sample Mendelian randomization when using heritable covariable-adjusted summary associations. Int. J. Epidemiol. 2021, 50, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Yki-Jarvinen, H.; Luukkonen, P.K. Heterogeneity of non-alcoholic fatty liver disease. Liver Int. 2015, 35, 2498–2500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Gill, D.; Butterworth, A.S.; Burgess, S. An empirical investigation into the impact of winner’s curse on estimates from Mendelian randomization. Int. J. Epidemiol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Munafò, M.R.; Tilling, K.; Taylor, A.E.; Evans, D.M.; Smith, G.D. Collider scope: When selection bias can substantially influence observed associations. Int. J. Epidemiol. 2018, 47, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Association for The Study of Liver Diseases (AASLD). The Liver Meeting: Pan-Society Presentation of NAFLD Nomenclature Consensus Process. 2023. Available online: https://www.youtube.com/watch?v=x29hpOSx620 (accessed on 20 January 2023).
- Kim, M.S.; Song, M.; Shin, J.I.; Won, H.H. How to interpret studies using Mendelian randomisation. BMJ Evid.-Based Med. 2023, in press. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Outcome | Method | Number of SNP | OR (95% CI) | p-Value |
|---|---|---|---|---|
| 25(OH)D vs. NAFLD | ||||
| Primary 1 | IVW-random effects | 6 | 0.95 (0.76–1.18) | 0.641 |
| MR Egger | 0.91 (0.59–1.39) | 0.684 | ||
| Weighted median | 0.97 (0.80–1.18) | 0.761 | ||
| Simple mode | 0.99 (0.69–1.41) | 0.951 | ||
| Weighted mode | 0.97 (0.78–1.20) | 0.790 | ||
| Sensitivity 1 | IVW-random effects | 6 | 1.04 (0.79–1.37) | 0.786 |
| MR Egger | 1.00 (0.59–1.71) | 0.990 | ||
| Weighted median | 1.03 (0.80–1.32) | 0.824 | ||
| Simple mode | 0.88 (0.55–1.41) | 0.612 | ||
| Weighted mode | 1.04 (0.79–1.35) | 0.801 | ||
| NAFLD vs. 25(OH)D | ||||
| Primary 2 | IVW-random effects | 5 | 1.00 (0.99–1.02) | 0.665 |
| MR Egger | 0.99 (0.96–1.02) | 0.523 | ||
| Weighted median | 1.00 (0.99–1.01) | 0.670 | ||
| Simple mode | 1.00 (0.99–1.01) | 0.789 | ||
| Weighted mode | 1.00 (0.99–1.01) | 0.701 | ||
| Sensitivity 2 | IVW-random effects | 4 | 1.00 (0.99–1.01) | 0.689 |
| MR Egger | 1.00 (0.96–1.04) | 0.951 | ||
| Weighted median | 1.00 (0.99–1.01) | 0.817 | ||
| Simple mode | 1.00 (0.98–1.02) | 0.960 | ||
| Weighted mode | 1.00 (0.99–1.01) | 0.763 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Burrows, K.; Fuller, H.; Speliotes, E.K.; Abeysekera, K.W.M.; Thorne, J.L.; Lewis, S.J.; Zulyniak, M.A.; Moore, J.B. Non-Alcoholic Fatty Liver Disease and Vitamin D in the UK Biobank: A Two-Sample Bidirectional Mendelian Randomisation Study. Nutrients 2023, 15, 1442. https://doi.org/10.3390/nu15061442
Zhang Z, Burrows K, Fuller H, Speliotes EK, Abeysekera KWM, Thorne JL, Lewis SJ, Zulyniak MA, Moore JB. Non-Alcoholic Fatty Liver Disease and Vitamin D in the UK Biobank: A Two-Sample Bidirectional Mendelian Randomisation Study. Nutrients. 2023; 15(6):1442. https://doi.org/10.3390/nu15061442
Chicago/Turabian StyleZhang, Zixuan, Kimberley Burrows, Harriett Fuller, Elizabeth K. Speliotes, Kushala W. M. Abeysekera, James L. Thorne, Sarah J. Lewis, Michael A. Zulyniak, and J. Bernadette Moore. 2023. "Non-Alcoholic Fatty Liver Disease and Vitamin D in the UK Biobank: A Two-Sample Bidirectional Mendelian Randomisation Study" Nutrients 15, no. 6: 1442. https://doi.org/10.3390/nu15061442
APA StyleZhang, Z., Burrows, K., Fuller, H., Speliotes, E. K., Abeysekera, K. W. M., Thorne, J. L., Lewis, S. J., Zulyniak, M. A., & Moore, J. B. (2023). Non-Alcoholic Fatty Liver Disease and Vitamin D in the UK Biobank: A Two-Sample Bidirectional Mendelian Randomisation Study. Nutrients, 15(6), 1442. https://doi.org/10.3390/nu15061442