
Clinical and Functional Assessment of Digenicity in Renal Phosphate Wasting

Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Recruitment and Study Protocol
2.2. In Vitro Studies
3. Results
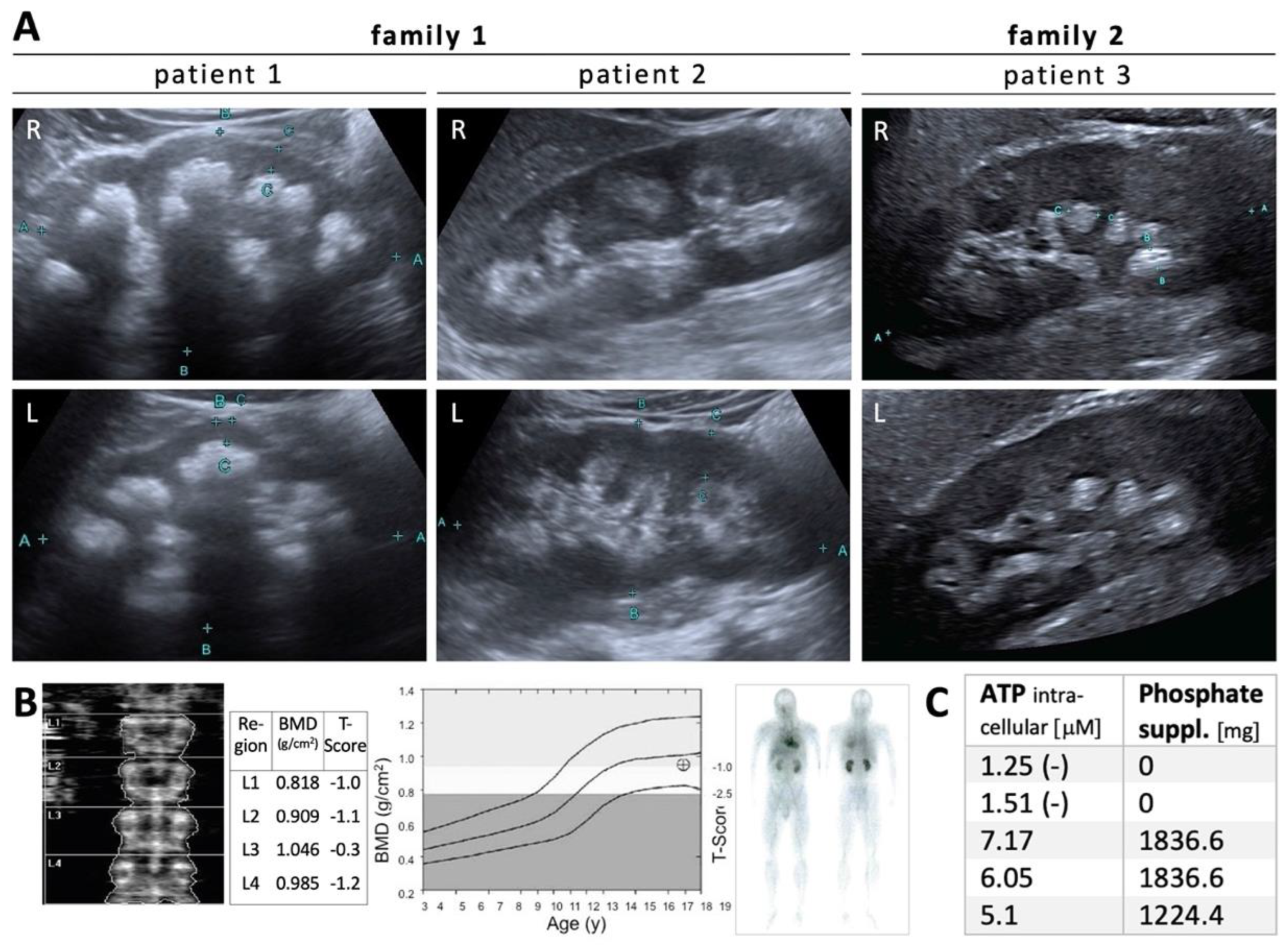
3.1. Phenotypic Characterization
3.2. Genotypic Characterization
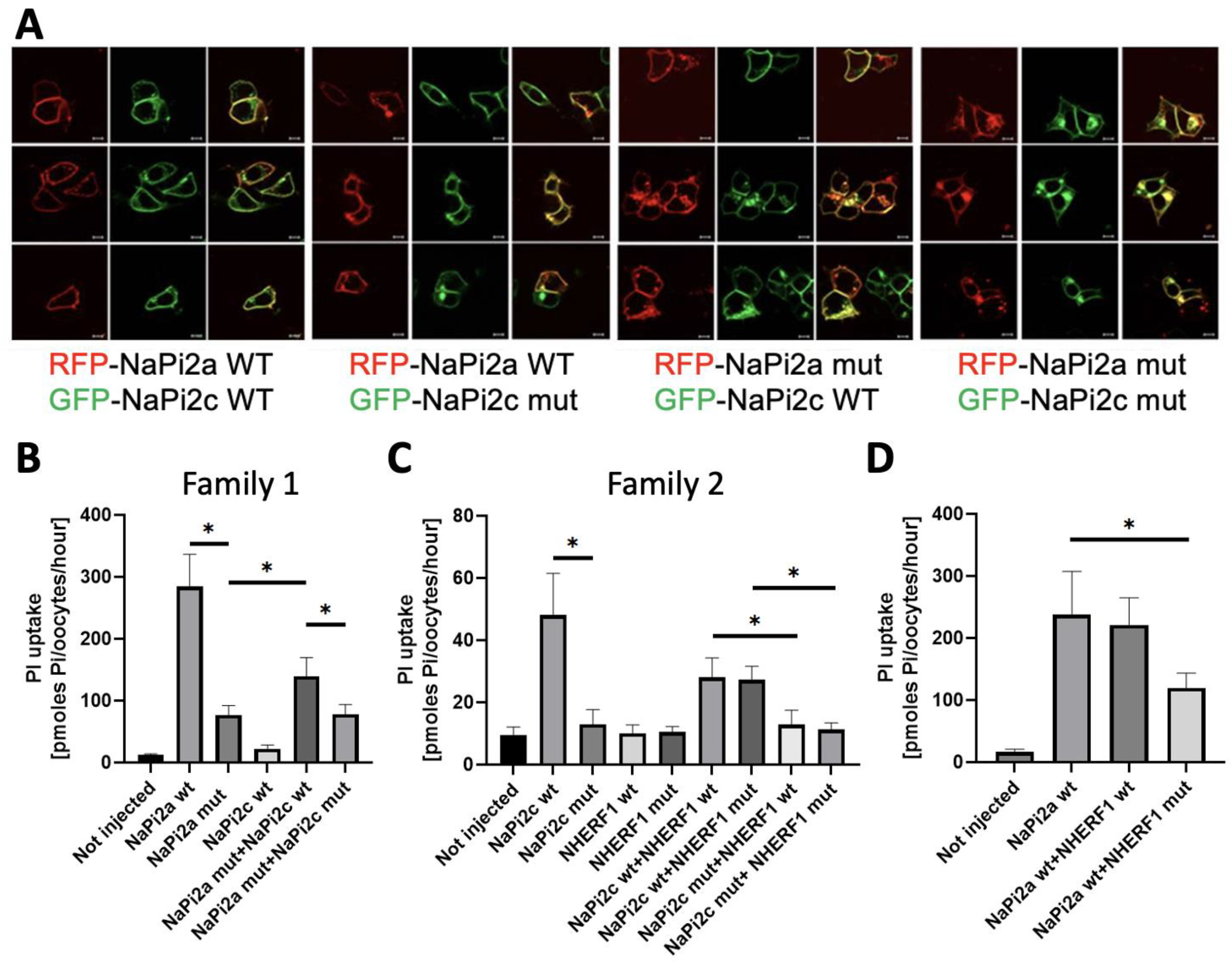
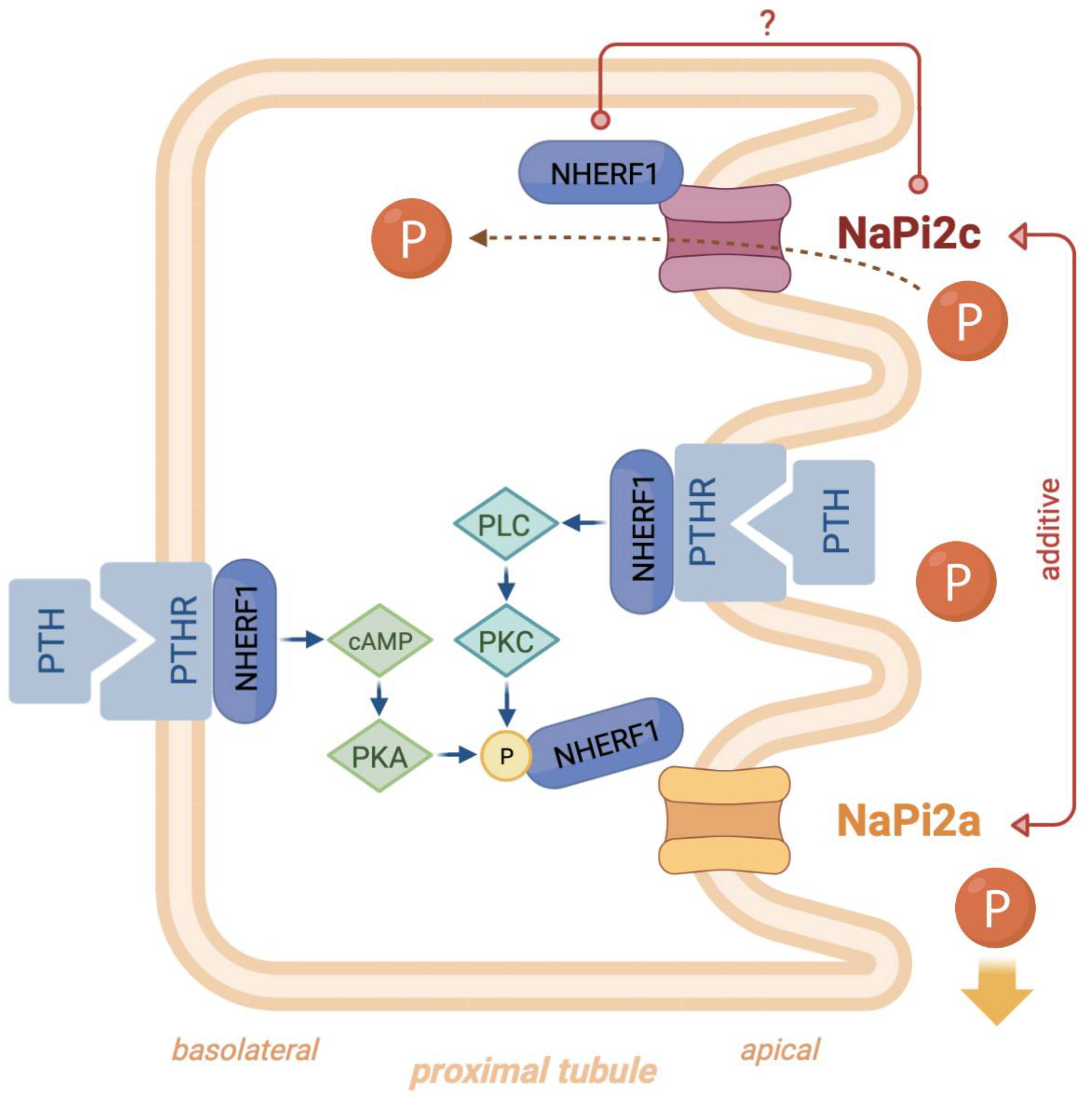
3.3. Functional Characterization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scales, C.D.; Smith, A.C.; Hanley, J.M.; Saigal, C.S. Prevalence of Kidney Stones in the United States. Eur. Urol. 2012, 62, 160–165. [Google Scholar] [CrossRef]
- Meneses, J.A.; Lucas, F.M.; Assunção, F.C.; Castro, J.P.P.; Monteiro, R.B. The Impact of Metaphylaxis of Kidney Stone Disease in the Renal Function at Long Term in Active Kidney Stone Formers Patients. Urol. Res. 2012, 40, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.; Zhu, W.; Robertson, W.G.; Penniston, K.L.; Smith, D.; Pozdzik, A.; Tefik, T.; Prezioso, D.; Pearle, M.S.; Chew, B.H.; et al. International Alliance of Urolithiasis (IAU) Guidelines on the Metabolic Evaluation and Medical Management of Urolithiasis. Urolithiasis 2022, 51, 4. [Google Scholar] [CrossRef]
- Tenenhouse, H.S.; Martel, J.; Gauthier, C.; Segawa, H.; Miyamoto, K. Differential Effects of Npt2a Gene Ablation and X-Linked Hyp Mutation on Renal Expression of Npt2c. Am. J. Physiol.-Renal Physiol. 2003, 285, F1271–F1278. [Google Scholar] [CrossRef] [PubMed]
- Ohkido, I.; Segawa, H.; Yanagida, R.; Nakamura, M.; Miyamoto, K. Cloning, Gene Structure and Dietary Regulation of the Type-IIc Na/Pi Cotransporter in the Mouse Kidney. Pflug. Arch 2003, 446, 106–115. [Google Scholar] [CrossRef]
- Lötscher, M.; Kaissling, B.; Biber, J.; Murer, H.; Levi, M. Role of Microtubules in the Rapid Regulation of Renal Phosphate Transport in Response to Acute Alterations in Dietary Phosphate Content. J. Clin. Investig. 1997, 99, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Mahon, M.J.; Donowitz, M.; Yun, C.C.; Segre, G.V. Na+/H+ Exchanger Regulatory Factor 2 Directs Parathyroid Hormone 1 Receptor Signalling. Nature 2002, 417, 858–861. [Google Scholar] [CrossRef]
- Courbebaisse, M.; Leroy, C.; Bakouh, N.; Salaün, C.; Beck, L.; Grandchamp, B.; Planelles, G.; Hall, R.A.; Friedlander, G.; Prié, D. A New Human NHERF1 Mutation Decreases Renal Phosphate Transporter NPT2a Expression by a PTH-Independent Mechanism. PLoS ONE 2012, 7, e34764. [Google Scholar] [CrossRef] [PubMed]
- Weinman, E.J.; Cunningham, R.; Wade, J.B.; Shenolikar, S. The Role of NHERF-1 in the Regulation of Renal Proximal Tubule Sodium-Hydrogen Exchanger 3 and Sodium-Dependent Phosphate Cotransporter 2a: NHERF-1 and Regulation of Renal Proximal Tubule NHE3 and Npt2a. J. Physiol. 2005, 567, 27–32. [Google Scholar] [CrossRef]
- Capuano, P.; Bacic, D.; Roos, M.; Gisler, S.M.; Stange, G.; Biber, J.; Kaissling, B.; Weinman, E.J.; Shenolikar, S.; Wagner, C.A.; et al. Defective Coupling of Apical PTH Receptors to Phospholipase C Prevents Internalization of the Na+-Phosphate Cotransporter NaPi-IIa in Nherf1-Deficient Mice. Am. J. Physiol.-Cell Physiol. 2007, 292, C927–C934. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Barac-Nieto, M.; Breusegem, S.Y.; Barry, N.P.; Levi, M.; Sorribas, V. Interactions of the Growth-Related, Type IIc Renal Sodium/Phosphate Cotransporter with PDZ Proteins. Kidney Int. 2008, 73, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Nowik, M.; Picard, N.; Stange, G.; Capuano, P.; Tenenhouse, H.S.; Biber, J.; Murer, H.; Wagner, C.A. Renal Phosphaturia during Metabolic Acidosis Revisited: Molecular Mechanisms for Decreased Renal Phosphate Reabsorption. Pflug. Arch. 2008, 457, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, X.; Wu, A. Association between a Novel Mutation in SLC20A2 and Familial Idiopathic Basal Ganglia Calcification. PLoS ONE 2013, 8, e57060. [Google Scholar] [CrossRef] [PubMed]
- Karim, Z.; Gérard, B.; Bakouh, N.; Alili, R.; Leroy, C.; Beck, L.; Silve, C.; Planelles, G.; Urena-Torres, P.; Grandchamp, B.; et al. NHERF1 Mutations and Responsiveness of Renal Parathyroid Hormone. N. Engl. J. Med. 2008, 359, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.J.; Li, D.; Doyle, D.; Zaritsky, J.; Levine, M.A. Digenic Heterozygous Mutations in SLC34A3 and SLC34A1 Cause Dominant Hypophosphatemic Rickets with Hypercalciuria. J. Clin. Endocrinol. Metab. 2020, 105, 2392–2400. [Google Scholar] [CrossRef]
- Brazier, F.; Courbebaisse, M.; David, A.; Bergerat, D.; Leroy, C.; Lindner, M.; Maruani, G.; Saint Jacques, C.; Letavernier, E.; Hureaux, M.; et al. Relationship between Clinical Phenotype and in Vitro Analysis of 13 NPT2c/SCL34A3 Mutants. Sci. Rep. 2023, 13, 85. [Google Scholar] [CrossRef]
- Mencarelli, M.A.; Heidet, L.; Storey, H.; van Geel, M.; Knebelmann, B.; Fallerini, C.; Miglietti, N.; Antonucci, M.F.; Cetta, F.; Sayer, J.A.; et al. Evidence of Digenic Inheritance in Alport Syndrome. J. Med. Genet. 2015, 52, 163–174. [Google Scholar] [CrossRef]
- Font-Llitjós, M.; Jiménez-Vidal, M.; Bisceglia, L.; Di Perna, M.; de Sanctis, L.; Rousaud, F.; Zelante, L.; Palacín, M.; Nunes, V. New Insights into Cystinuria: 40 New Mutations, Genotype-Phenotype Correlation, and Digenic Inheritance Causing Partial Phenotype. J. Med. Genet. 2005, 42, 58–68. [Google Scholar] [CrossRef]
- Perea-Romero, I.; Solarat, C.; Blanco-Kelly, F.; Sanchez-Navarro, I.; Bea-Mascato, B.; Martin-Salazar, E.; Lorda-Sanchez, I.; Swafiri, S.T.; Avila-Fernandez, A.; Martin-Merida, I.; et al. Allelic Overload and Its Clinical Modifier Effect in Bardet-Biedl Syndrome. NPJ Genom. Med. 2022, 7, 41. [Google Scholar] [CrossRef]
- Schäffer, A.A. Digenic Inheritance in Medical Genetics. J. Med. Genet. 2013, 50, 641–652. [Google Scholar] [CrossRef]
- Halbritter, J.; Seidel, A.; Müller, L.; Schönauer, R.; Hoppe, B. Update on Hereditary Kidney Stone Disease and Introduction of a New Clinical Patient Registry in Germany. Front. Pediatr. 2018, 6, 47. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Schönauer, R.; Petzold, F.; Lucinescu, W.; Seidel, A.; Müller, L.; Neuber, S.; Bergmann, C.; Sayer, J.A.; Werner, A.; Halbritter, J. Evaluating Pathogenicity of SLC34A3-Ser192Leu, a Frequent European Missense Variant in Disorders of Renal Phosphate Wasting. Urolithiasis 2019, 47, 511–519. [Google Scholar] [CrossRef]
- Fearn, A.; Allison, B.; Rice, S.J.; Edwards, N.; Halbritter, J.; Bourgeois, S.; Pastor-Arroyo, E.M.; Hildebrandt, F.; Tasic, V.; Wagner, C.A.; et al. Clinical, Biochemical, and Pathophysiological Analysis of SLC34A1 Mutations. Physiol. Rep. 2018, 6, e13715. [Google Scholar] [CrossRef] [PubMed]
- Markovich, D. Expression Cloning and Radiotracer Uptakes in Xenopus Laevis Oocytes. Nat. Protoc. 2008, 3, 1975–1980. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh Naderi, A.S.; Reilly, R.F. Hereditary Disorders of Renal Phosphate Wasting. Nat. Rev. Nephrol. 2010, 6, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Edmonston, D.; Wolf, M. FGF23 at the Crossroads of Phosphate, Iron Economy and Erythropoiesis. Nat. Rev. Nephrol. 2020, 16, 7–19. [Google Scholar] [CrossRef]
- Wolf, M.; Rubin, J.; Achebe, M.; Econs, M.J.; Peacock, M.; Imel, E.A.; Thomsen, L.L.; Carpenter, T.O.; Weber, T.; Brandenburg, V.; et al. Effects of Iron Isomaltoside vs Ferric Carboxymaltose on Hypophosphatemia in Iron-Deficiency Anemia: Two Randomized Clinical Trials. JAMA 2020, 323, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Schlingmann, K.P.; Ruminska, J.; Kaufmann, M.; Dursun, I.; Patti, M.; Kranz, B.; Pronicka, E.; Ciara, E.; Akcay, T.; Bulus, D.; et al. Autosomal-Recessive Mutations in SLC34A1 Encoding Sodium-Phosphate Cotransporter 2A Cause Idiopathic Infantile Hypercalcemia. JASN 2016, 27, 604–614. [Google Scholar] [CrossRef]
- Schönauer, R.; Scherer, L.; Nemitz-Kliemchen, M.; Hagemann, T.; Hantmann, E.; Seidel, A.; Müller, L.; Kehr, S.; Voigt, C.; Stolzenburg, J.-U.; et al. Systematic Assessment of Monogenic Etiology in Adult-Onset Kidney Stone Formers Undergoing Urological Intervention-Evidence for Genetic Pretest Probability. Am. J. Med. Genet. C Semin. Med. Genet. 2022, 190, 279–288. [Google Scholar] [CrossRef]
- Sun, B.B.; Kurki, M.I.; Foley, C.N.; Mechakra, A.; Chen, C.-Y.; Marshall, E.; Wilk, J.B.; Biogen Biobank Team; Chahine, M.; Chevalier, P.; et al. Genetic Associations of Protein-Coding Variants in Human Disease. Nature 2022, 603, 95–102. [Google Scholar] [CrossRef]
- Bieri, C.; Daryadel, A.; Bettoni, C.; Pastor-Arroyo, E.-M.; Schnitzbauer, U.; Hernando, N.; Wagner, C.A. The Human Pathogenic 91del7 Mutation in SLC34A1 Has No Effect in Mineral Homeostasis in Mice. Sci. Rep. 2022, 12, 6102. [Google Scholar] [CrossRef] [PubMed]
- Mamonova, T.; Friedman, P.A. Noncanonical Sequences Involving NHERF1 Interaction with NPT2A Govern Hormone-Regulated Phosphate Transport: Binding Outside the Box. Int. J. Mol. Sci. 2021, 22, 1087. [Google Scholar] [CrossRef]
- Hernando, N.; Gisler, S.M.; Pribanic, S.; Déliot, N.; Capuano, P.; Wagner, C.A.; Moe, O.W.; Biber, J.; Murer, H. NaPi-IIa and Interacting Partners. J. Physiol. 2005, 567, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Shenolikar, S.; Voltz, J.W.; Minkoff, C.M.; Wade, J.B.; Weinman, E.J. Targeted Disruption of the Mouse NHERF-1 Gene Promotes Internalization of Proximal Tubule Sodium-Phosphate Cotransporter Type IIa and Renal Phosphate Wasting. Proc. Natl. Acad. Sci. USA 2002, 99, 11470–11475. [Google Scholar] [CrossRef]
- Mamonova, T.; Kurnikova, M.; Friedman, P.A. Structural Basis for NHERF1 PDZ Domain Binding. Biochemistry 2012, 51, 3110–3120. [Google Scholar] [CrossRef] [PubMed]
- Pesta, D.H.; Tsirigotis, D.N.; Befroy, D.E.; Caballero, D.; Jurczak, M.J.; Rahimi, Y.; Cline, G.W.; Dufour, S.; Birkenfeld, A.L.; Rothman, D.L.; et al. Hypophosphatemia Promotes Lower Rates of Muscle ATP Synthesis. FASEB J. 2016, 30, 3378–3387. [Google Scholar] [CrossRef]
- Felsenfeld, A.J.; Levine, B.S. Approach to Treatment of Hypophosphatemia. Am. J. Kidney Dis. 2012, 60, 655–661. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fam 1, Pat 1 | Fam 1, Pat 2 | Fam 2, Pat 3 | |
|---|---|---|---|
| Nephrocalcinosis/kidney stone disease | Severe nephrocalcinosis, recurrent kidney stones | Mild nephrocalcinosis | Mild nephrocalcinosis |
| Bone disease | Increased bone metabolism, short stature | No | Stress fracture tibia |
| Other symptoms | No | No | Muscle weakness/pain, tachycardia |
| Phosphate supplement | Yes | No | Yes |
| Sex | Male | Female | Female |
| Age (time of phenotype exploration) | 18 | 47 | 51 |
| Digenic variants, ACMG classification | - Hom. SLC34A3 p.(Ser192Leu); P (PS3, PM1, PM2, PP5) - Hom. SLC34A1 p.(Val91_Ala97del); VUS (PS3, BS1) | - Het. SLC34A3 p.(Ser192Leu); P (PS3, PM1,PM2,PP5) - Het. SLC34A1 p.(Val91_Ala97del); VUS (PS3,BS1) | - Het. SLC34A3 p.(Ser192Leu); P (PS3, PM1,PM2,PP5) - Het. NHERF1 p.(Glu225Lys); LP (PS3, PM1, BP4) |
| eGFR (mL/min/1.73 m2) | 82 (−) | 100 | 56 (−) |
| Serum phosphate (mmol/L) | 0.63 (−) | 1.11 | 0.45 (−) |
| FGF-23 C-terminal (RU/mL) | 36 | 835 (+) | 62 |
| TrP (%) | 77 (−) | 88 | 68.5 (−) |
| TmP/GFR (mmol/L) | 0.49 (−) | 0.98 | 0.69 (−) |
| Serum calcium (mmol/L) | 2.68 (+) | 2.40 | 2.51 |
| Urine calcium (Ca/Crea ratio in mmol/mmol Crea) | 0.23 | 0.54 | 0.23 |
| Serum PTH (pmol/L) | 0.80 (−) | 2.12 | 2.88 |
| 1.25 (OH)2 vit. D3 (pg/mL) | 44.0 | 70.7 | 54.8 |
| 25(OH) vit. D3 (ng/mL) | 17.2 (−) | 25.8 | 24.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petzold, F.; Schönauer, R.; Werner, A.; Halbritter, J. Clinical and Functional Assessment of Digenicity in Renal Phosphate Wasting. Nutrients 2023, 15, 2081. https://doi.org/10.3390/nu15092081
Petzold F, Schönauer R, Werner A, Halbritter J. Clinical and Functional Assessment of Digenicity in Renal Phosphate Wasting. Nutrients. 2023; 15(9):2081. https://doi.org/10.3390/nu15092081
Chicago/Turabian StylePetzold, Friederike, Ria Schönauer, Andreas Werner, and Jan Halbritter. 2023. "Clinical and Functional Assessment of Digenicity in Renal Phosphate Wasting" Nutrients 15, no. 9: 2081. https://doi.org/10.3390/nu15092081
APA StylePetzold, F., Schönauer, R., Werner, A., & Halbritter, J. (2023). Clinical and Functional Assessment of Digenicity in Renal Phosphate Wasting. Nutrients, 15(9), 2081. https://doi.org/10.3390/nu15092081