

The Short-Term Variation of Human Gut Mycobiome in Response to Dietary Intervention of Different Macronutrient Distributions


Abstract
:1. Introduction
2. Materials and Methods
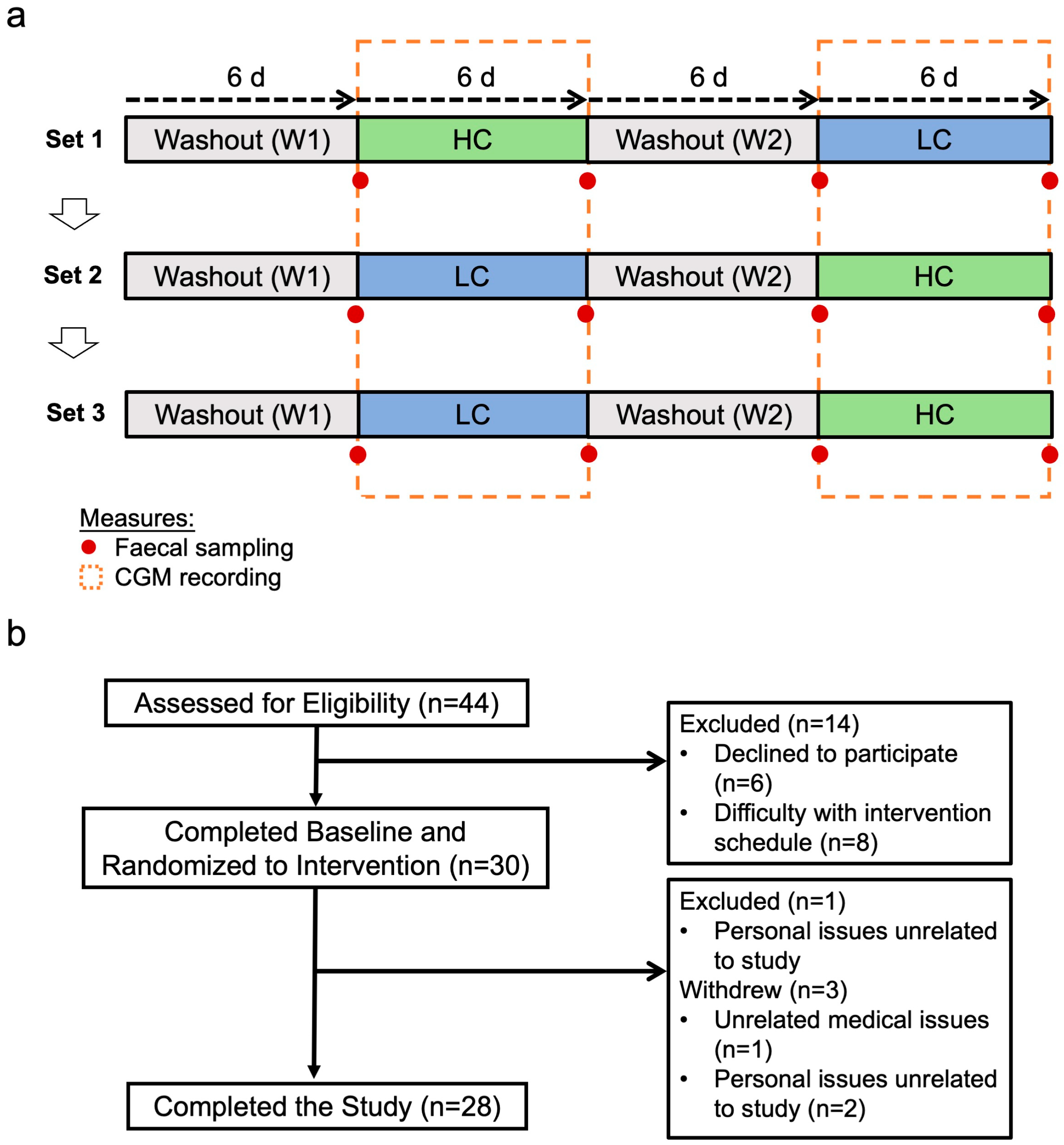
2.1. Study Design and Participants
2.2. DNA Extraction and Sequencing
2.3. Bioinformatic Analysis
2.4. Statistical Analysis
3. Results
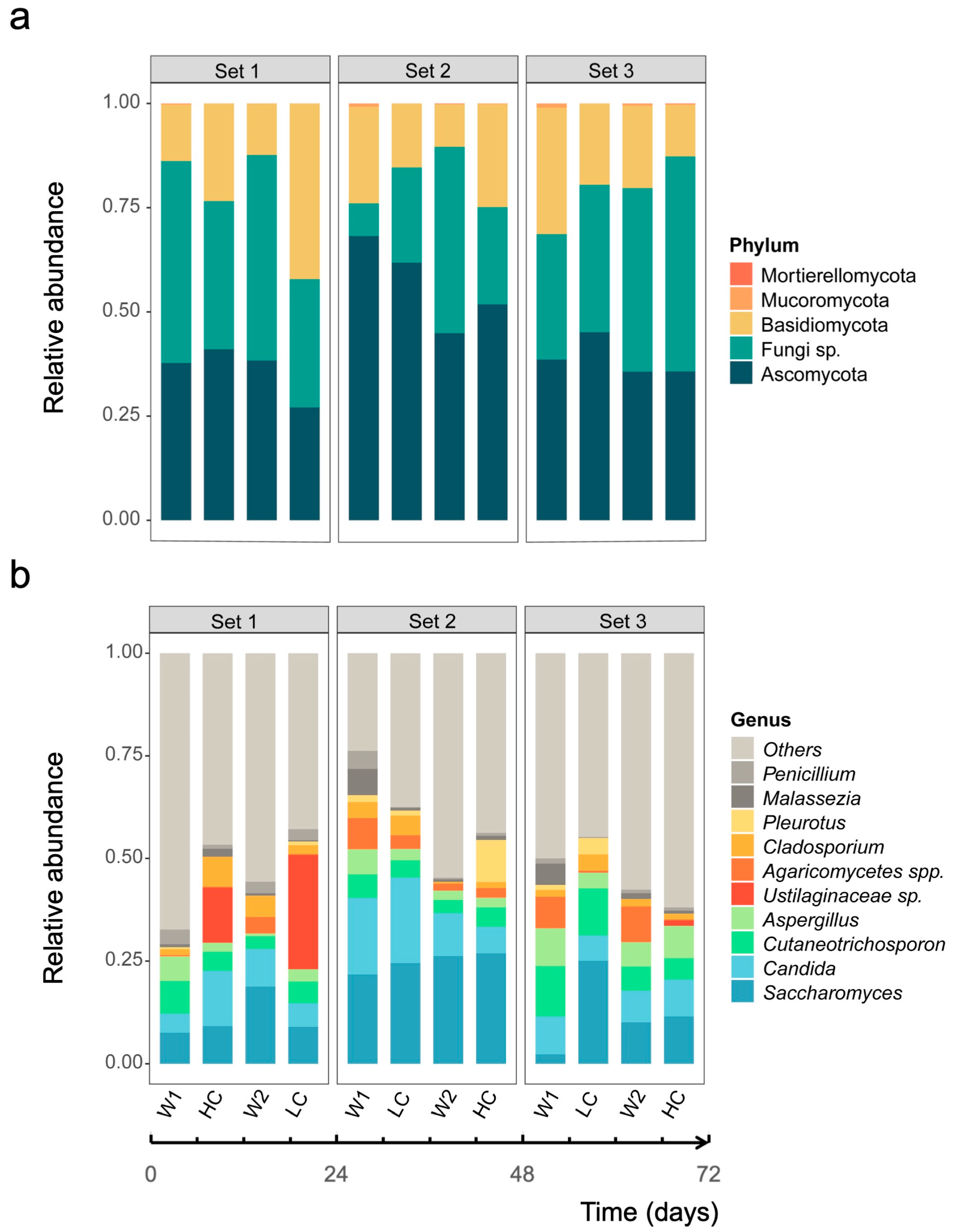
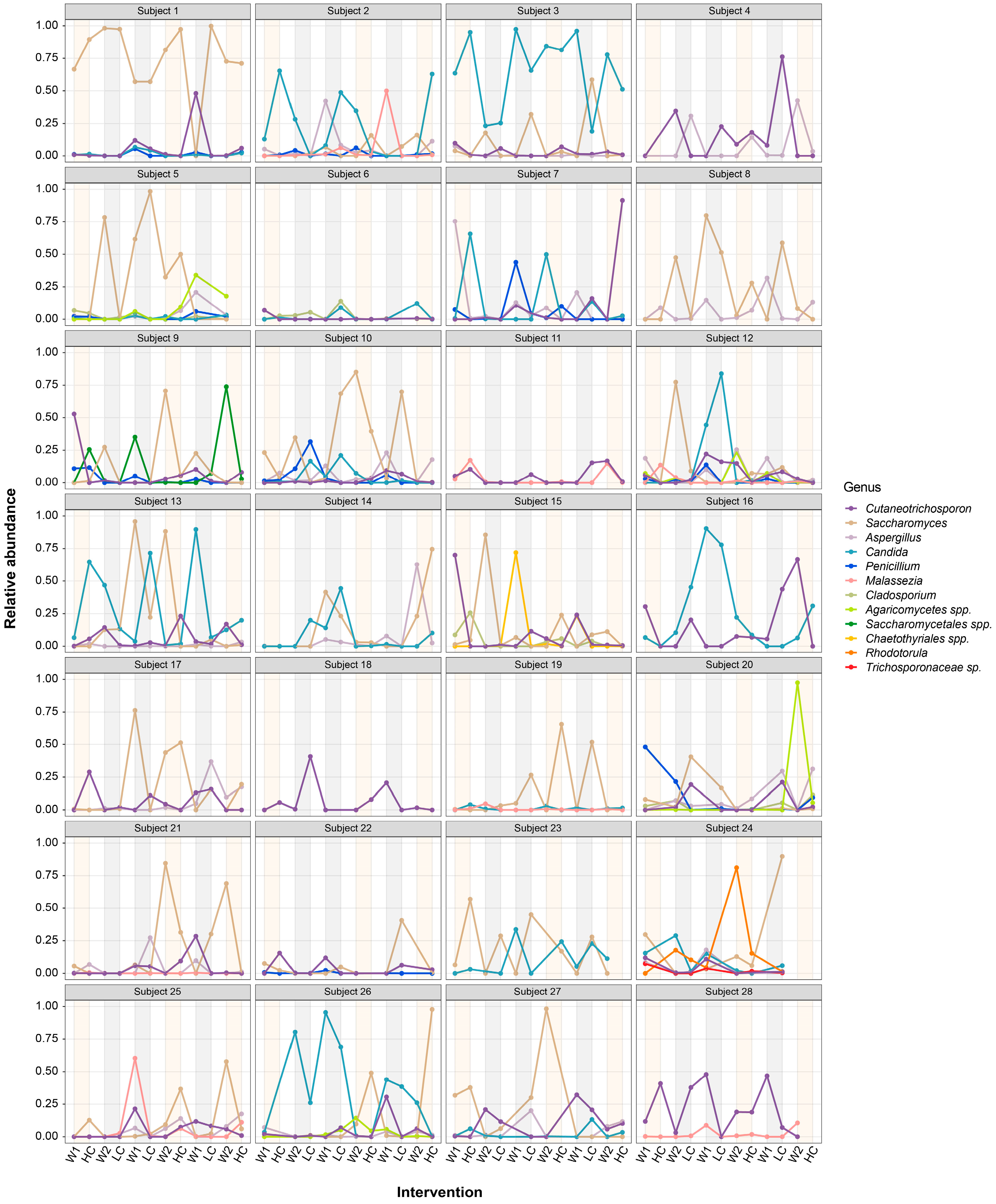
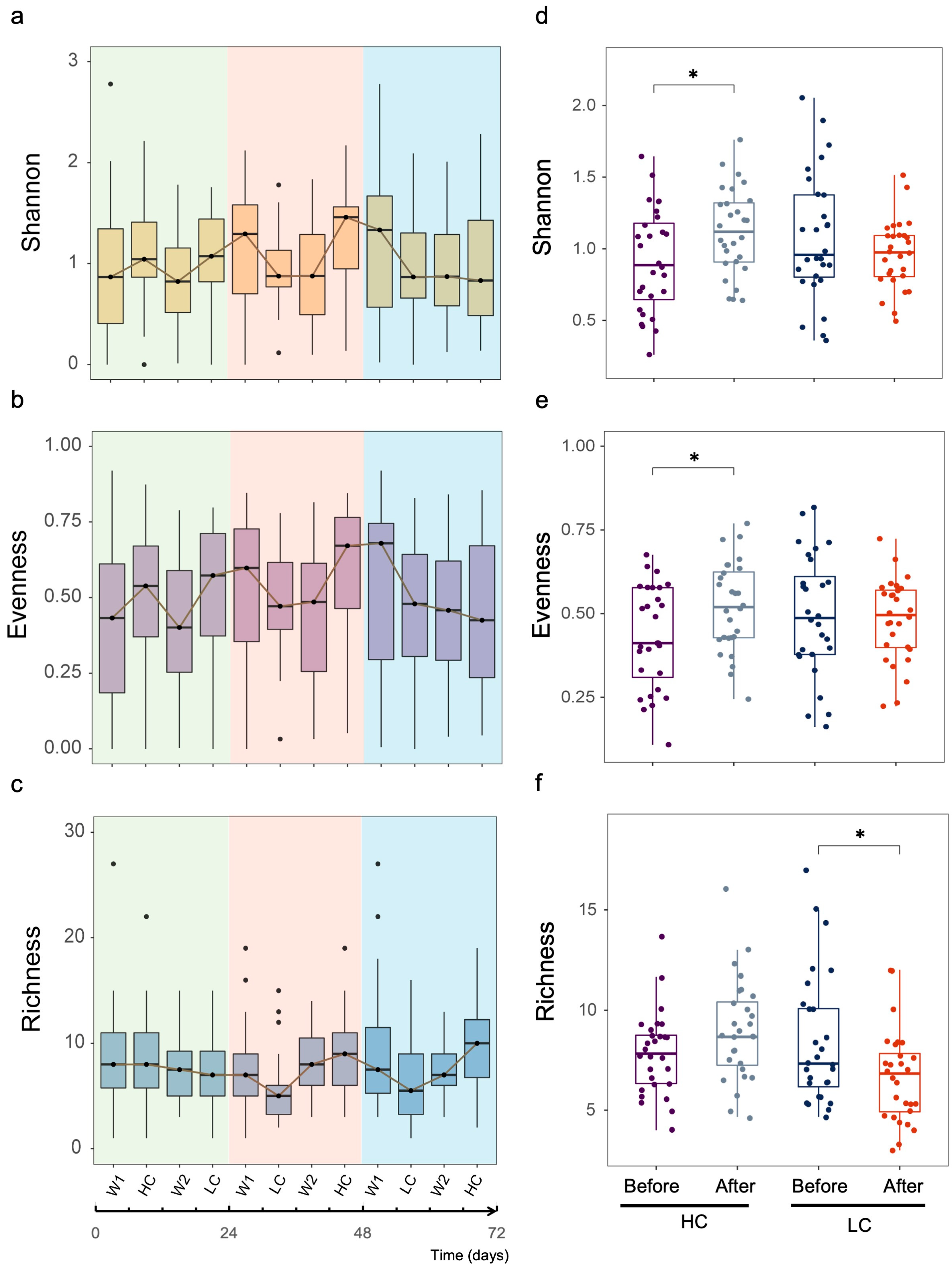
3.1. Community Profiles of Gut Mycobiome over Study Period
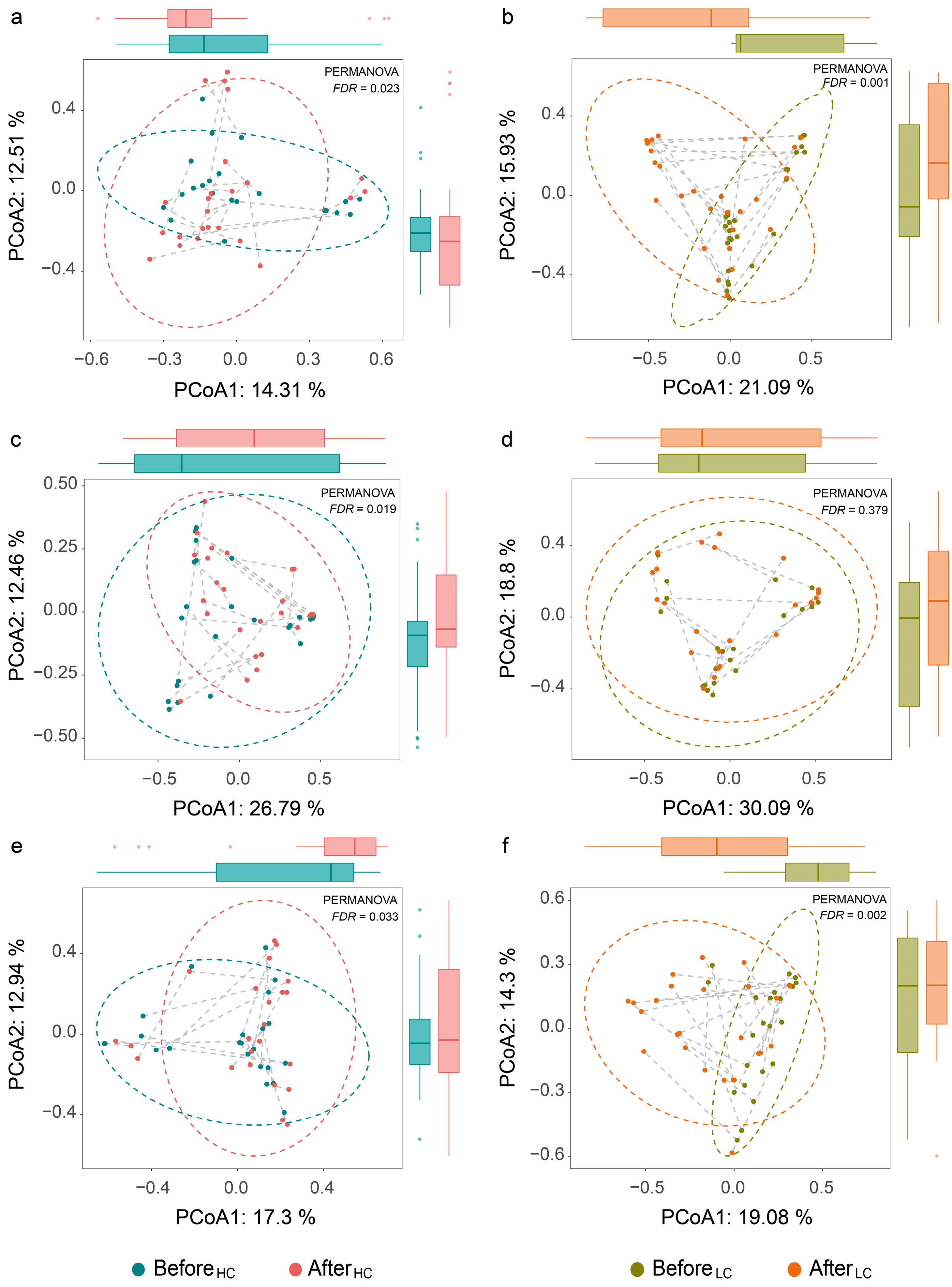
3.2. Dynamic Changes in Human Gut Mycobiome Composition Induced by Dietary Intervention
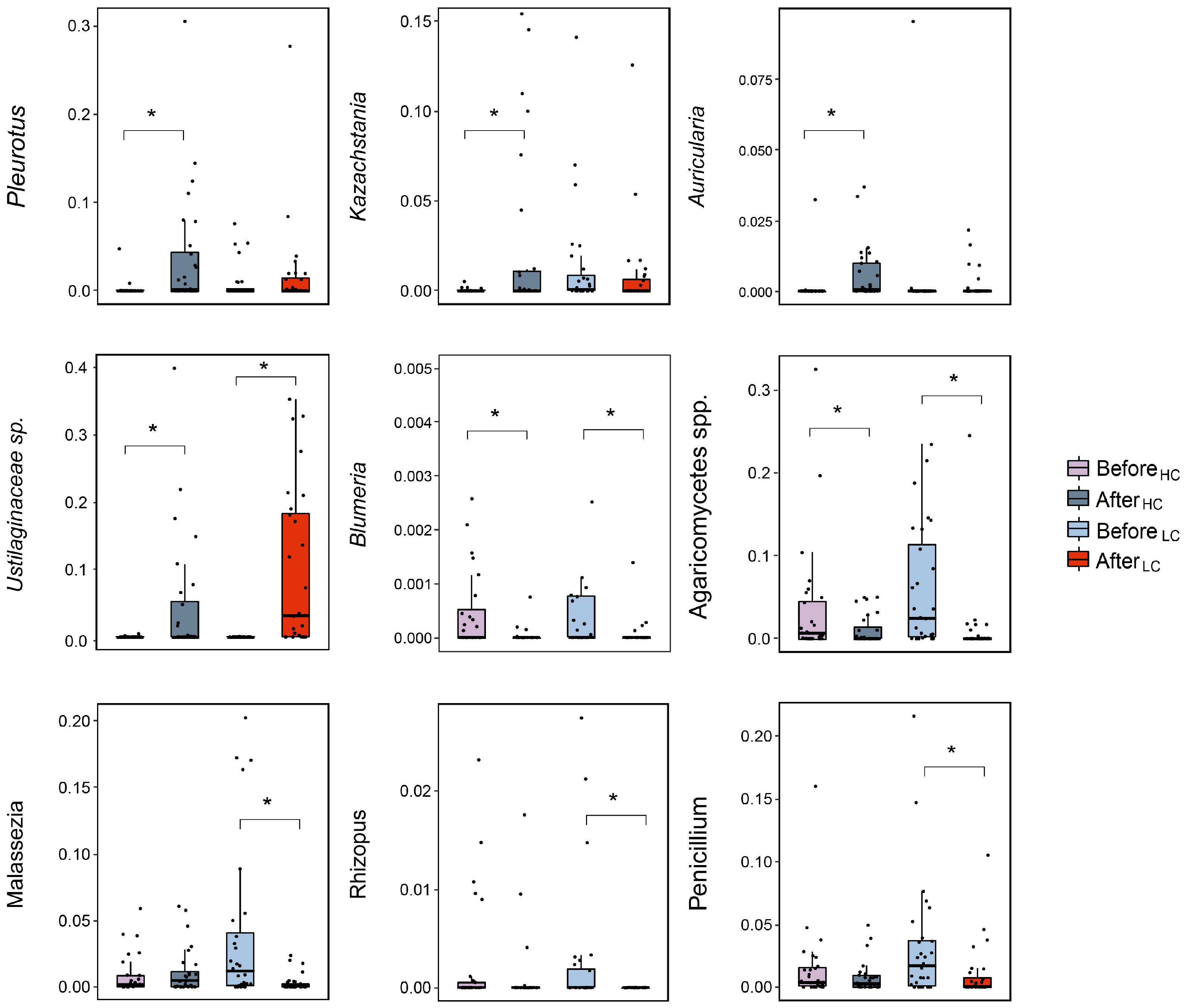
3.3. Differential Effects between HC and LC Dietary Interventions on Gut Fungal Composition
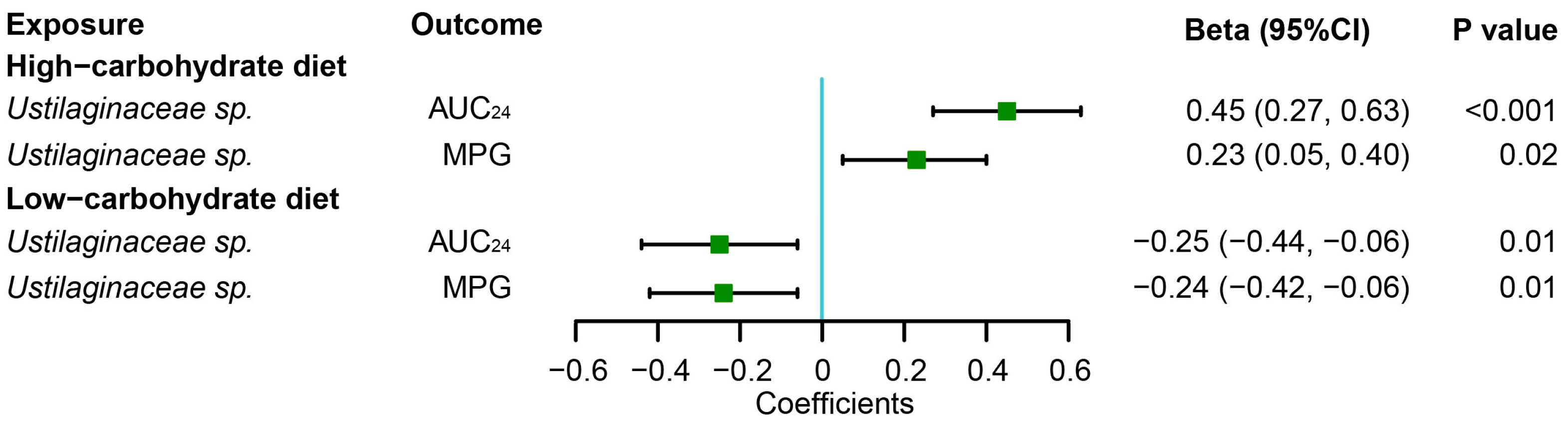
3.4. Associations of Gut Fungi with Postprandial Glycemic Responses to Dietary Intervention
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bäckhed, F.; Fraser, C.M.; Ringel, Y.; Sanders, M.E.; Sartor, R.B.; Sherman, P.M.; Versalovic, J.; Young, V.; Finlay, B.B. Defining a healthy human gut microbiome: Current concepts, future directions, and clinical applications. Cell Host Microbe 2012, 12, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Iliev, I.D. The mycobiota: Interactions between commensal fungi and the host immune system. Nat. Rev. Immunol. 2014, 14, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, D.; Yu, B.; He, J.; Zheng, P.; Mao, X.; Yu, J.; Luo, J.; Tian, G.; Huang, Z.; et al. Fungi in Gastrointestinal Tracts of Human and Mice: From Community to Functions. Microb. Ecol. 2018, 75, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Gosiewski, T.; Salamon, D.; Szopa, M.; Sroka, A.; Malecki, M.T.; Bulanda, M. Quantitative evaluation of fungi of the genus Candida in the feces of adult patients with type 1 and 2 diabetes—A pilot study. Gut Pathog. 2014, 6, 43. [Google Scholar] [CrossRef]
- Huffnagle, G.B.; Noverr, M.C. The emerging world of the fungal microbiome. Trends Microbiol. 2013, 21, 334–341. [Google Scholar] [CrossRef]
- Zhang, L.; Zhan, H.; Xu, W.; Yan, S.; Ng, S.C. The role of gut mycobiome in health and diseases. Ther. Adv. Gastroenterol. 2021, 14, 1–18. [Google Scholar] [CrossRef]
- Ott, S.J.; Kühbacher, T.; Musfeldt, M.; Rosenstiel, P.; Hellmig, S.; Rehman, A.; Drews, O.; Weichert, W.; Timmis, K.N.; Schreiber, S. Fungi and inflammatory bowel diseases: Alterations of composition and diversity. Scand. J. Gastroenterol 2008, 43, 831–841. [Google Scholar] [CrossRef]
- Luan, C.; Xie, L.; Yang, X.; Miao, H.; Lv, N.; Zhang, R.; Xiao, X.; Hu, Y.; Liu, Y.; Wu, N.; et al. Dysbiosis of fungal microbiota in the intestinal mucosa of patients with colorectal adenomas. Sci. Rep. 2015, 5, 7980. [Google Scholar] [CrossRef]
- Shankar, J. Food Habit Associated Mycobiota Composition and Their Impact on Human Health. Front. Nutr. 2021, 8, 773577. [Google Scholar] [CrossRef]
- Graf, D.; Di Cagno, R.; Fåk, F.; Flint, H.J.; Nyman, M.; Saarela, M.; Watzl, B. Contribution of diet to the composition of the human gut microbiota. Microb. Ecol. Health Dis. 2015, 26, 26164. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Ukhanova, M.; Wang, X.; Baer, D.J.; Novotny, J.A.; Fredborg, M.; Mai, V. Effects of almond and pistachio consumption on gut microbiota composition in a randomised cross-over human feeding study. Br. J. Nutr. 2014, 111, 2146–2152. [Google Scholar] [CrossRef]
- Auchtung, T.A.; Fofanova, T.Y.; Stewart, C.J.; Nash, A.K.; Wong, M.C.; Gesell, J.R.; Auchtung, J.M.; Ajami, N.J.; Petrosino, J.F. Investigating Colonization of the Healthy Adult Gastrointestinal Tract by Fungi. mSphere 2018, 3, e00092-18. [Google Scholar] [CrossRef] [PubMed]
- Pareek, S.; Kurakawa, T.; Das, B.; Motooka, D.; Nakaya, S.; Rongsen-Chandola, T.; Goyal, N.; Kayama, H.; Dodd, D.; Okumura, R.; et al. Comparison of Japanese and Indian intestinal microbiota shows diet-dependent interaction between bacteria and fungi. NPJ Biofilms Microbiomes 2019, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Fu, Y.; Tian, Y.; Gou, W.; Miao, Z.; Yang, M.; Ordovas, J.M.; Zheng, J.S. Individual Postprandial Glycemic Responses to Diet in n-of-1 Trials: Westlake N-of-1 Trials for Macronutrient Intake (WE-MACNUTR). J. Nutr. 2021, 151, 3158–3167. [Google Scholar] [CrossRef]
- Tian, Y.; Ma, Y.; Fu, Y.; Zheng, J.S. Application of n-of-1 Clinical Trials in Personalized Nutrition Research: A Trial Protocol for Westlake N-of-1 Trials for Macronutrient Intake (WE-MACNUTR). Curr. Dev. Nutr. 2020, 4, nzaa143. [Google Scholar] [CrossRef]
- Hamad, S.; Ranque, E.I.; Azhar, M.; Yasir, A.; Jiman-Fatani, A.; Tissot-Dupont, H.; Raoult, D.; Bittar, F. Culturomics and Amplicon-based Metagenomic Approaches for the Study of Fungal Population in Human Gut Microbiota. Sci. Rep. 2017, 7, 16788. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. A Guide Methods Appl. 1990, 18, 315–322. [Google Scholar]
- Shuai, M.; Fu, Y.; Zhong, H.L.; Gou, W.; Jiang, Z.; Liang, Y.; Miao, Z.; Xu, J.J.; Huynh, T.; Wahlqvist, M.L.; et al. Mapping the human gut mycobiome in middle-aged and elderly adults: Multiomics insights and implications for host metabolic health. Gut 2022, 71, 1812–1820. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Schulze, J.; Sonnenborn, U. Yeasts in the gut: From commensals to infectious agents. Dtsch Arztebl. Int. 2009, 106, 837–842. [Google Scholar] [CrossRef]
- Nash, K.; Auchtung, T.A.; Wong, M.C.; Smith, D.P.; Gesell, J.R.; Ross, M.C.; Stewart, C.J.; Metcalf, G.A.; Muzny, D.M.; Gibbs, R.A.; et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 2017, 5, 153. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.L.; Sokol, H. The gut mycobiota: Insights into analysis, environmental interactions and role in gastrointestinal diseases. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 331–345. [Google Scholar] [CrossRef]
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and fungi of the human gut microbiome: Correlations with diet and bacterial residents. PLoS ONE 2013, 8, e66019. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.M.; Perez, D.; Chaves, F.J.; Esteve, E.; Marin-Garcia, P.; Xifra, G.; Vendrell, J.; Jove, M.; Pamplona, R.; Ricart, W.; et al. Obesity changes the human gut mycobiome. Sci. Rep. 2015, 5, 14600. [Google Scholar] [CrossRef]
- Hallen-Adams, H.E.; Suhr, M.J. Fungi in the healthy human gastrointestinal tract. Virulence 2017, 8, 352–358. [Google Scholar] [CrossRef]
- Sun, Y.; Zuo, T.; Cheung, C.P.; Gu, W.; Wan, Y.; Zhang, F.; Chen, N.; Zhan, H.; Yeoh, Y.K.; Niu, J.; et al. Population-Level Configurations of Gut Mycobiome Across 6 Ethnicities in Urban and Rural China. Gastroenterology 2021, 160, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Heisel, T.; Montassier, E.; Johnson, A.; Al-Ghalith, G.; Lin, Y.W.; Wei, L.N.; Knights, D.; Gale, C.A. High-Fat Diet Changes Fungal Microbiomes and Interkingdom Relationships in the Murine Gut. mSphere 2017, 2, e00351-17. [Google Scholar] [CrossRef] [PubMed]
- Jayasuriya, W.J.; Wanigatunge, C.A.; Fernando, G.H.; Abeytunga, D.T.; Suresh, T.S. Hypoglycaemic activity of culinary Pleurotus ostreatus and P. cystidiosus mushrooms in healthy volunteers and type 2 diabetic patients on diet control and the possible mechanisms of action. Phytother. Res. 2015, 29, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Xu, J.; Sheng, Y.; Liu, J.; Li, H.; Guo, M.; Xu, W.; Luo, Y.; Huang, K.; He, X. Pleurotus Ostreatus Ameliorates Obesity by Modulating the Gut Microbiota in Obese Mice Induced by High-Fat Diet. Nutrients 2022, 14, 1868. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Chen, X.; Song, J.; Chen, M.; Gong, P.; Jia, W.; Li, G. Hypoglycemic effects of Auricularia auricula polysaccharides on high fat diet and streptozotocin-induced diabetic mice using metabolomics analysis. Food Funct. 2021, 12, 9994–10007. [Google Scholar] [CrossRef] [PubMed]
- Korcari, D.; Ricci, G.; Capusoni, C.; Fortina, M.G. Physiological performance of Kazachstania unispora in sourdough environments. World J. Microbiol. Biotechnol. 2021, 37, 88. [Google Scholar] [CrossRef]
- Carbonetto, B.; Nidelet, T.; Guezenec, S.; Perez, M.; Segond, D.; Sicard, D. Interactions between Kazachstania humilis Yeast Species and Lactic Acid Bacteria in Sourdough. Microorganisms 2020, 8, 240. [Google Scholar] [CrossRef]
- Suhr, M.J.; Banjara, N.; Hallen-Adams, H.E. Sequence-based methods for detecting and evaluating the human gut mycobiome. Lett. Appl. Microbiol. 2016, 62, 209–215. [Google Scholar] [CrossRef]
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci Transl. Med. 2012, 4, 165rv113. [Google Scholar] [CrossRef]
- Denning, D.W.; Bromley, M.J. Infectious Disease. How to bolster the antifungal pipeline. Science 2015, 347, 1414–1416. [Google Scholar] [CrossRef] [PubMed]
- Strati, F.; Di Paola, M.; Stefanini, I.; Albanese, D.; Rizzetto, L.; Lionetti, P.; Calabrò, A.; Jousson, O.; Donati, C.; Cavalieri, D.; et al. Age and Gender Affect the Composition of Fungal Population of the Human Gastrointestinal Tract. Front. Microbiol. 2016, 7, 1227. [Google Scholar] [CrossRef]
- Cui, L.; Morris, A.; Ghedin, E. The human mycobiome in health and disease. Genome Med. 2013, 5, 63. [Google Scholar] [CrossRef]
- Hallen-Adams, H.E.; Kachman, S.D.; Kim, J.; Legge, R.M.; Martínez, I. Fungi inhabiting the healthy human gastrointestinal tract: A diverse and dynamic community. Fungal Ecol. 2015, 15, 9–17. [Google Scholar] [CrossRef]
- Zhang, F.; Aschenbrenner, D.; Yoo, J.Y.; Zuo, T. The gut mycobiome in health, disease, and clinical applications in association with the gut bacterial microbiome assembly. Lancet Microbe 2022, 3, e969–e983. [Google Scholar] [CrossRef] [PubMed]
- Paulino, B.N.; Pessôa, M.G.; Molina, G.; Neto, A.A.K.; Oliveira, J.V.C.; Mano, M.C.R.; Pastore, G.M. Biotechnological production of value-added compounds by ustilaginomycetous yeasts. Appl. Microbiol. Biotechnol. 2017, 101, 7789–7809. [Google Scholar] [CrossRef] [PubMed]
- Ullmann, L.; Phan, A.N.T.; Kaplan, D.K.P.; Blank, L.M. Ustilaginaceae Biocatalyst for Co-Metabolism of CO(2)-Derived Substrates toward Carbon-Neutral Itaconate Production. J. Fungi 2021, 7, 98. [Google Scholar] [CrossRef]
- Ullmann, L.; Wibberg, D.; Busche, T.; Rückert, C.; Müsgens, A.; Kalinowski, J.; Blank, L.M. Seventeen Ustilaginaceae High-Quality Genome Sequences Allow Phylogenomic Analysis and Provide Insights into Secondary Metabolite Synthesis. J. Fungi 2022, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Wardman, J.F.; Bains, R.K.; Rahfeld, P.; Withers, S.G. Carbohydrate-active enzymes (CAZymes) in the gut microbiome. Nat. Rev. Microbiol. 2022, 20, 542–556. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Overall (n = 28) | |
|---|---|
| Age (years) | 25.8 ± 2.7 |
| BMI (kg/m2) | 22.1 ± 3.0 |
| Waist circumference (cm) | 78.9 ± 9.1 |
| Current drinking (%) | |
| Occasionally | 6 (67) |
| Never | 3 (33) |
| Fasting clinical parameters | |
| Insulin (mU/L) | 25.4 ± 5.3 |
| Fasting plasma glucose (mmol/L) | 4.20 ± 0.26 |
| Triglycerides (mmol/L) | 0.77 ± 0.38 |
| Total cholesterol (mmol/L) | 4.30 ± 0.77 |
| LDL cholesterol (mmol/L) | 1.92 ± 0.59 |
| HDL cholesterol (mmol/L) | 1.67 ± 0.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Y.; Gou, W.; Ma, Y.; Shuai, M.; Liang, X.; Fu, Y.; Zheng, J.-S. The Short-Term Variation of Human Gut Mycobiome in Response to Dietary Intervention of Different Macronutrient Distributions. Nutrients 2023, 15, 2152. https://doi.org/10.3390/nu15092152
Tian Y, Gou W, Ma Y, Shuai M, Liang X, Fu Y, Zheng J-S. The Short-Term Variation of Human Gut Mycobiome in Response to Dietary Intervention of Different Macronutrient Distributions. Nutrients. 2023; 15(9):2152. https://doi.org/10.3390/nu15092152
Chicago/Turabian StyleTian, Yunyi, Wanglong Gou, Yue Ma, Menglei Shuai, Xinxiu Liang, Yuanqing Fu, and Ju-Sheng Zheng. 2023. "The Short-Term Variation of Human Gut Mycobiome in Response to Dietary Intervention of Different Macronutrient Distributions" Nutrients 15, no. 9: 2152. https://doi.org/10.3390/nu15092152
APA StyleTian, Y., Gou, W., Ma, Y., Shuai, M., Liang, X., Fu, Y., & Zheng, J. -S. (2023). The Short-Term Variation of Human Gut Mycobiome in Response to Dietary Intervention of Different Macronutrient Distributions. Nutrients, 15(9), 2152. https://doi.org/10.3390/nu15092152