
Molecular Mechanism of Calycosin Inhibited Vascular Calcification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Lentiviral Transduction
2.2. Reagents and Antibodies
2.3. Calcium Content and Alkaline Phosphatase (ALP) Activity
2.4. Alizarin Red S Staining
2.5. Protein Extraction and Western Blotting
2.6. Immunoprecipitation
2.7. Autophagic Flux Assay
2.8. Bovine Serum Albumin (BSA) Dequenching Assay
2.9. LysoTracker Assay
2.10. Statistical Analyses
3. Results
3.1. Calycosin Treatment Inhibits β-GP-Induced Osteogenic Differentiation and Calcification in A7r5 Cells In Vitro
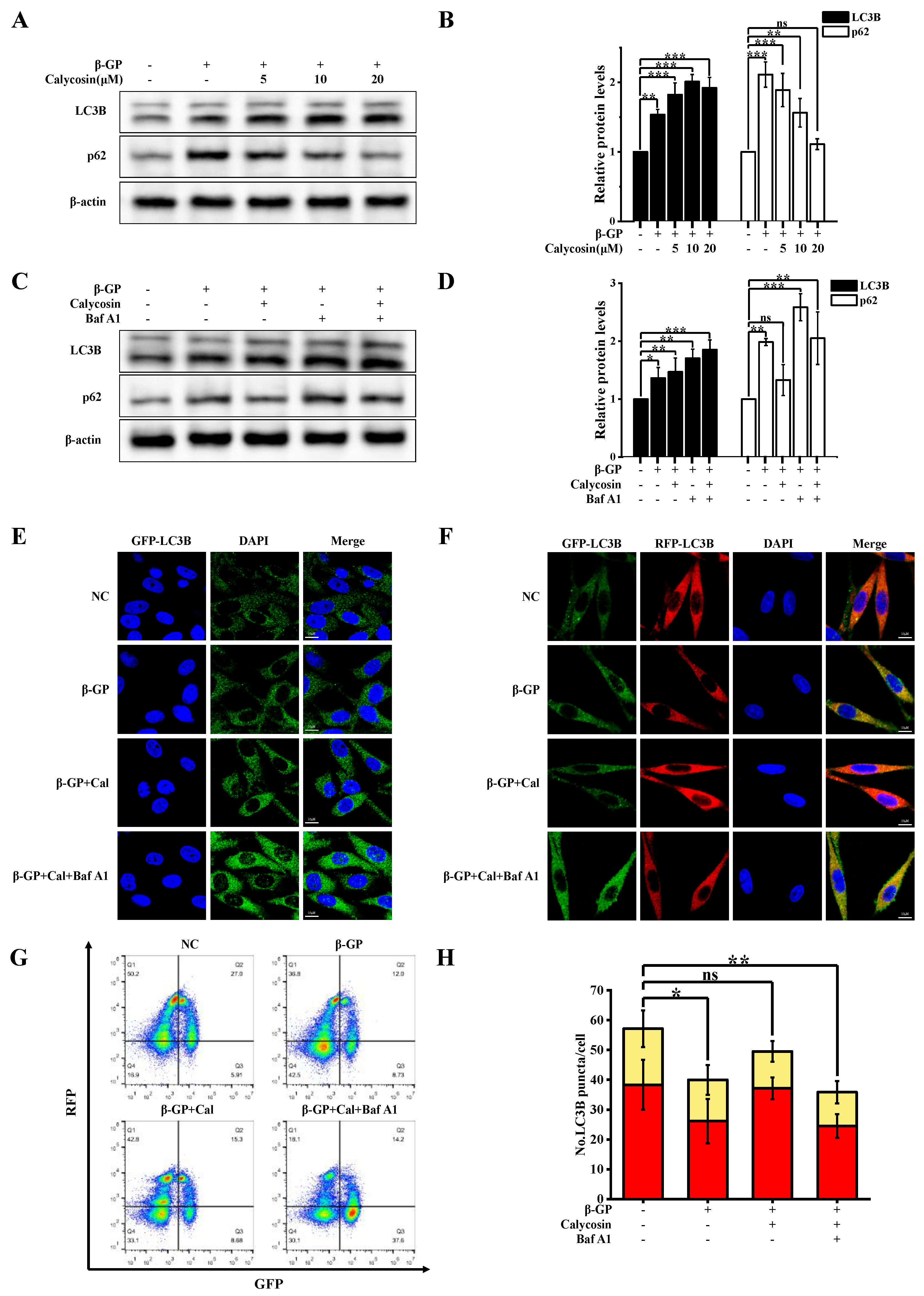
3.2. Calycosin Increases Autophagic Flux, but Has No Effect on Lysosome Function in Calcified Cells
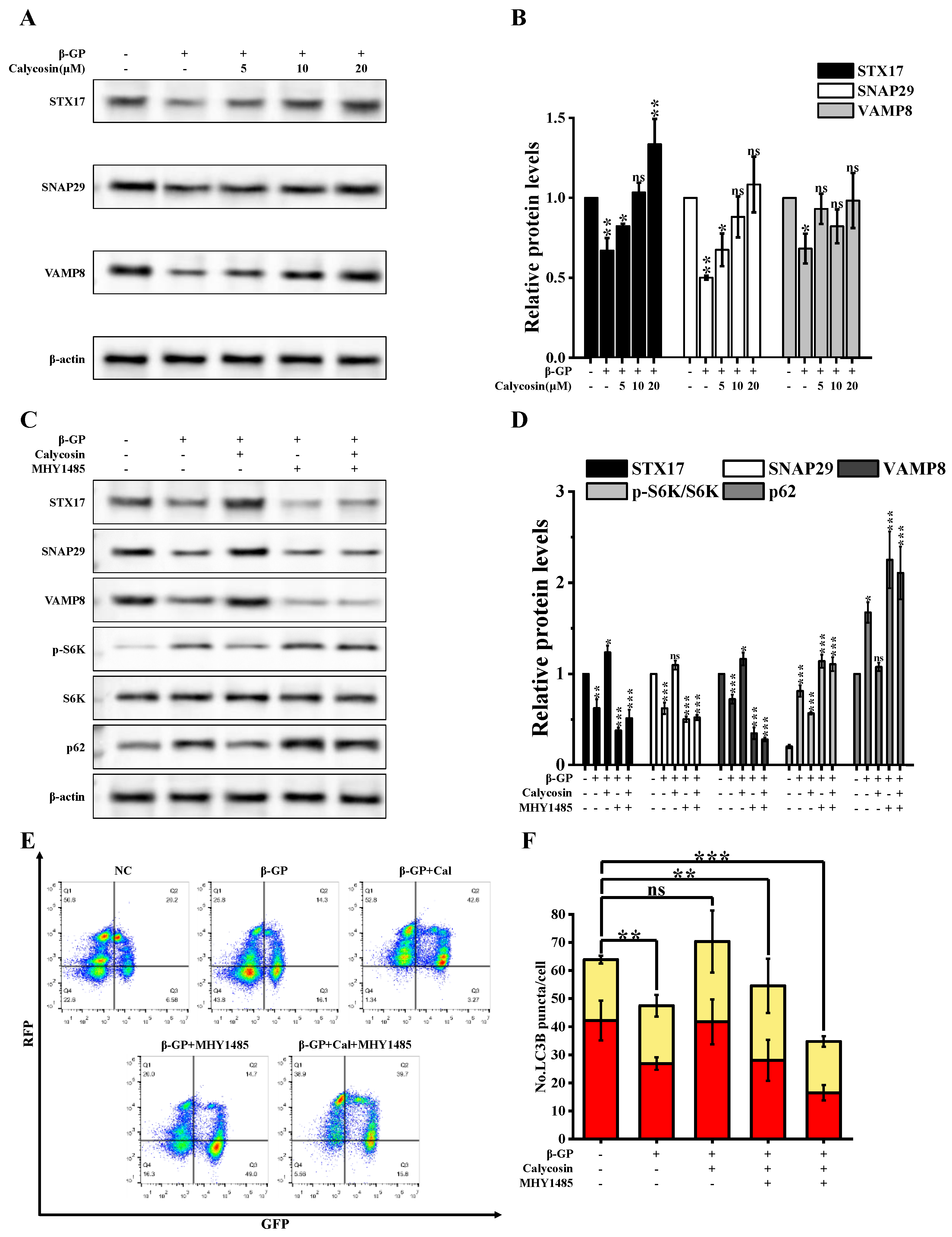
3.3. Calycosin Initiates Autophagy via Activation of AMPK/mTOR Signaling Pathway in Calcified Cells
3.4. Calycosin Inhibits Calcification by Promoting SNARE Complex-Mediated Autophagosome–Lysosome Fusion
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, S.J.; Lee, I.-K.; Jeon, J.-H. Vascular Calcification-New Insights into Its Mechanism. Int. J. Mol. Sci. 2020, 21, 2685. [Google Scholar] [CrossRef] [PubMed]
- Vervloet, M.; Cozzolino, M. Vascular calcification in chronic kidney disease: Different bricks in the wall? Kidney Int. 2017, 91, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Yoo, T.-K.; Lee, S.-H.; Rhim, H.-C.; Lee, M.-Y.; Cheong, E.-S.; Seo, M.-H.; Sung, K.-C. Association of Cardiovascular Mortality with Concurrent Coronary Artery Calcification and Physical Activity: A Cohort Study. Med. Lith. 2023, 59, 522. [Google Scholar] [CrossRef] [PubMed]
- Bellasi, A.; Di Lullo, L.; Russo, D.; Ciarcia, R.; Magnocavallo, M.; Lavalle, C.; Ratti, C.; Cozzolino, M.; Di Iorio, B.R. Vascular Calcification Progression Modulates the Risk Associated with Vascular Calcification Burden in Incident to Dialysis Patients. Cells 2021, 10, 1091. [Google Scholar] [CrossRef] [PubMed]
- Steitz, S.A.; Speer, M.Y.; Curinga, G.; Yang, H.Y.; Haynes, P.; Aebersold, R.; Schinke, T.; Karsenty, G.; Giachelli, C.M. Smooth muscle cell phenotypic transition associated with calcification—Upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ. Res. 2001, 89, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.C.; Leopold, J.A.; Loscalzo, J. Vascular calcification—Pathobiological mechanisms and clinical implications. Circ. Res. 2006, 99, 1044–1059. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; McKee, M.D.; Steitz, S.; Giachelli, C.M. Calcification of vascular smooth muscle cell cultures inhibition by osteopontin. Circ. Res. 1999, 84, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xiang, P.; Chen, M.; Luol, Y.; Zhao, Y.; Zhu, J.; Jing, W.; Yu, H. Nano-Sized Hydroxyapatite Induces Apoptosis and Osteogenic Differentiation of Vascular Smooth Muscle Cells via JNK/c-JUN Pathway. Int. J. Nanomed. 2021, 16, 3633–3648. [Google Scholar] [CrossRef]
- Liu, Q.; Luo, Y.; Zhao, Y.; Xiang, P.; Zhu, J.; Jing, W.; Jin, W.; Chen, M.; Tang, R.; Yu, H. Nano-hydroxyapatite accelerates vascular calcification via lysosome impairment and autophagy dysfunction in smooth muscle cells. Bioact. Mater. 2022, 8, 478–493. [Google Scholar] [CrossRef]
- Cao, J.; Chen, C.; Chen, Q.; Gao, Y.; Zhao, Z.; Yuan, Q.; Li, A.; Yang, S.; He, Y.; Zu, X.; et al. Extracellular vesicle miR-32 derived from macrophage promotes arterial calcification in mice with type 2 diabetes via inhibiting VSMC autophagy. J. Transl. Med. 2022, 20, 307. [Google Scholar] [CrossRef]
- Dai, X.-Y.; Zhao, M.-M.; Cai, Y.; Guan, Q.-C.; Zhao, Y.; Guan, Y.; Kong, W.; Zhu, W.-G.; Xu, M.-J.; Wang, X. Phosphate-induced autophagy counteracts vascular calcification by reducing matrix vesicle release. Kidney Int. 2013, 83, 1042–1051. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, J.; Meng, Q.; Li, D.; Hu, F.-Z.; Zhu, Y.-Q.; Huang, Y.-Y.; Liu, Y.-N.; Sun, L.; Liang, Q.-H. The protective effects of long non-coding RNA-ANCR on arterial calcification. J. Bone Miner. Metab. 2020, 38, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tan, S.-H.; Nicolas, V.; Bauvy, C.; Yang, N.-D.; Zhang, J.; Xue, Y.; Codogno, P.; Shen, H.-M. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 2013, 23, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Ouyang, Q.; Zhu, M.; Yu, H.; Mei, K.; Liu, R. mTOR-mediated phosphorylation of VAMP8 and SCFD1 regulates autophagosome maturation. Nat. Commun. 2021, 12, 6622. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Liu, G.; Ding, M.; Zong, G.; Hu, F.B.; Willett, W.C.; Rimm, E.B.; Manson, J.E.; Sun, Q. Isoflavone Intake and the Risk of Coronary Heart Disease in US Men and Women Results From 3 Prospective Cohort Studies. Circulation 2020, 141, 1127–1137. [Google Scholar] [CrossRef]
- Huang, F.; Chen, J.-Y.; Ouyang, J.-M. Comparison of the inhibition of high phosphate-induced smooth muscle cell calcification by Porphyra yezoensis and Astragalus polysaccharides. J. Funct. Foods 2020, 73, 104160. [Google Scholar] [CrossRef]
- Ma, C.; Wu, H.; Yang, G.; Xiang, J.; Feng, K.; Zhang, J.; Hua, Y.; Kang, L.; Fan, G.; Yang, S. Calycosin ameliorates atherosclerosis by enhancing autophagy via regulating the interaction between KLF2 and MLKL in apolipoprotein E gene-deleted mice. Br. J. Pharmacol. 2022, 179, 252–269. [Google Scholar] [CrossRef]
- Liu, C.; Wang, X.; Wang, X.; Zhang, Y.; Min, W.; Yu, P.; Miao, J.; Shen, W.; Chen, S.; Zhou, S.; et al. A new LKB1 activator, piericidin analogue S14, retards renal fibrosis through promoting autophagy and mitochondrial homeostasis in renal tubular cells. Theranostics 2022, 12, 7158–7179. [Google Scholar] [CrossRef]
- Liang, X.; Yu, C.; Tian, Y.; Xiang, X.; Luo, Y. Inhibition of STX17-SNAP29-VAMP8 complex formation by costunolide sensitizes ovarian cancer cells to cisplatin via the AMPK/mTOR signaling pathway. Biochem. Pharmacol. 2023, 212, 115549. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xu, S.-N.; Yuan, S.-T.; Lei, X.; Sun, X.; Xing, L.; Li, H.-J.; He, C.-X.; Qin, W.; Zhao, D.; et al. Multiple functions of autophagy in vascular calcification. Cell Biosci. 2021, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Lanzer, P.; Hannan, M.F.; Lanzer, D.J.; Janzen, J.; Raggi, P.; Furniss, D.; Schuchardt, M.; Thakker, R.; Fok, P.-W.; Saez-Rodriguez, J.; et al. Medial Arterial Calcification JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 1145–1165. [Google Scholar] [CrossRef] [PubMed]
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; St Hilaire, C.; Shanahan, C. Medial vascular calcification revisited: Review and perspectives. Eur. Heart J. 2014, 35, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Phadwal, K.; Feng, D.; Zhu, D.; MacRae, V.E. Autophagy as a novel therapeutic target in vascular calcification. Pharmacol. Ther. 2020, 206, 107430. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Lu, L.; Gao, L.; Wang, Y.; Wang, W. Calycosin attenuates doxorubicin-induced cardiotoxicity via autophagy regulation in zebrafish models. Biomed. Pharmacother. 2021, 137, 111375. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Huang, P.; Yang, J.; Du, H.; Wan, H.; He, Y. Calycosin alleviates cerebral ischemia/reperfusion injury by repressing autophagy via STAT3/FOXO3a signaling pathway. Phytomedicine 2023, 115, 154845. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. Embo J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Phadwal, K.; Koo, E.; Jones, R.A.; Forsythe, R.O.; Tang, K.; Tang, Q.; Corcoran, B.M.; Caporali, A.; MacRae, V.E. Metformin protects against vascular calcification through the selective degradation of Runx2 by the p62 autophagy receptor. J. Cell. Physiol. 2022, 237, 4303–4316. [Google Scholar] [CrossRef]
- Pang, Q.; Wang, P.; Pan, Y.; Dong, X.; Zhou, T.; Song, X.; Zhang, A. Irisin protects against vascular calcification by activating autophagy and inhibiting NLRP3-mediated vascular smooth muscle cell pyroptosis in chronic kidney disease. Cell Death Dis. 2022, 13, 283. [Google Scholar] [CrossRef]
- Li, F.-X.-Z.; Liu, J.-J.; Xu, F.; Shan, S.-K.; Zheng, M.-H.; Lei, L.-M.; Lin, X.; Guo, B.; Li, C.-C.; Wu, F.; et al. Cold exposure protects against medial arterial calcification development via autophagy. J. Nanobiotechnol. 2023, 21, 226. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Z.; Li, Y.; Jiang, W.; Wang, Z. Molecular Mechanism of Calycosin Inhibited Vascular Calcification. Nutrients 2024, 16, 99. https://doi.org/10.3390/nu16010099
Zhou Z, Li Y, Jiang W, Wang Z. Molecular Mechanism of Calycosin Inhibited Vascular Calcification. Nutrients. 2024; 16(1):99. https://doi.org/10.3390/nu16010099
Chicago/Turabian StyleZhou, Zekun, Yi Li, Wei Jiang, and Zengli Wang. 2024. "Molecular Mechanism of Calycosin Inhibited Vascular Calcification" Nutrients 16, no. 1: 99. https://doi.org/10.3390/nu16010099
APA StyleZhou, Z., Li, Y., Jiang, W., & Wang, Z. (2024). Molecular Mechanism of Calycosin Inhibited Vascular Calcification. Nutrients, 16(1), 99. https://doi.org/10.3390/nu16010099