The Potential Protective Action of Vitamin D in Hepatic Insulin Resistance and Pancreatic Islet Dysfunction in Type 2 Diabetes Mellitus

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Vitamin D Synthesis and Metabolism
3. Vitamin D and T2DM
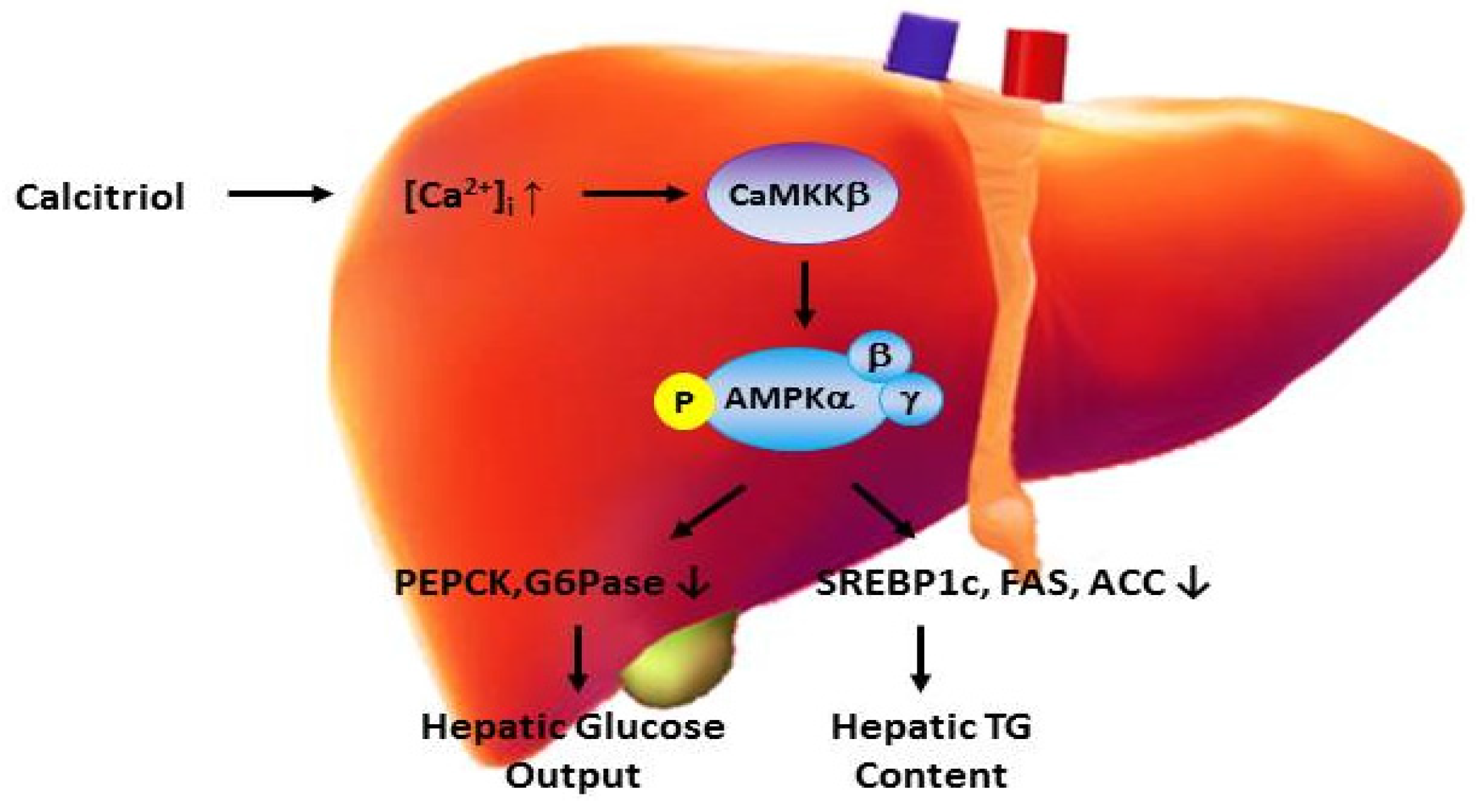
4. Vitamin D and Hepatic Metabolism
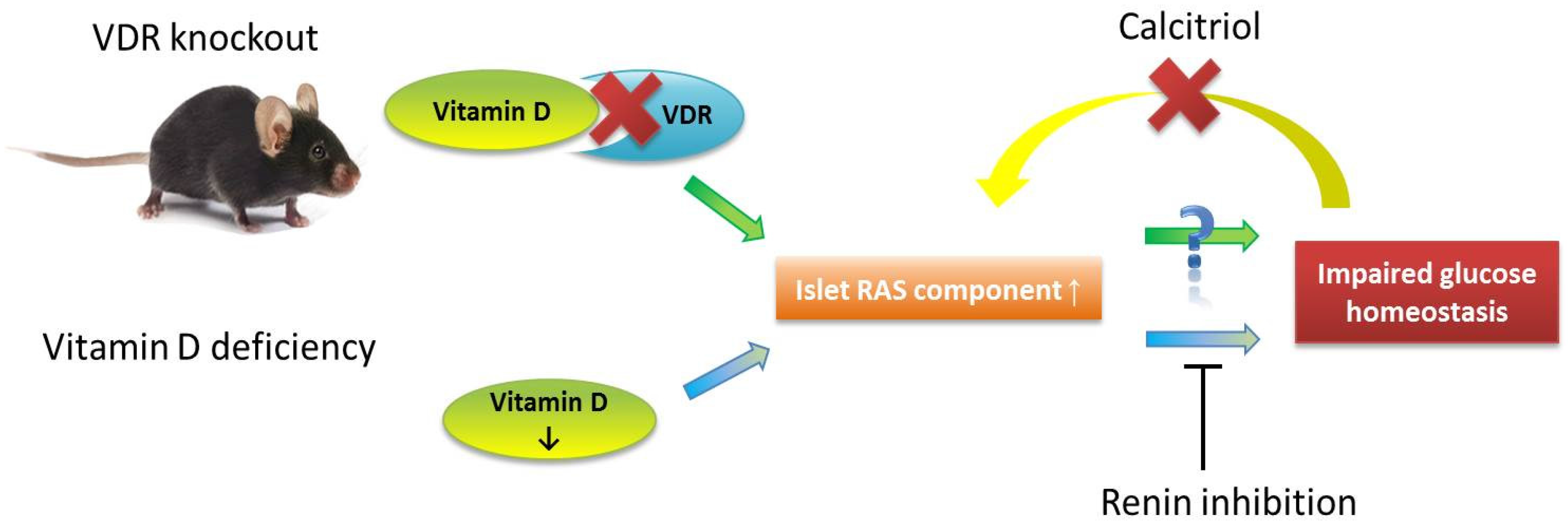
5. Vitamin D and Pancreatic Islet Function
6. Vitamin D and Pancreatic Islet Development
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Black, D.D. Hepatobiliary physiology. In The Gastrointestinal System; Leung, P.S., Ed.; Springer: Berlin, Germany, 2014; pp. 237–324. [Google Scholar]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [PubMed]
- Wallace, I.R.; Wallace, H.J.; Mckinley, M.C.; Bell, P.M.; Hunter, S.J. Vitamin D and insulin resistance. Clin. Endocrinol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Nwosu, B.U.; Maranda, L. The effects of vitamin D supplementation on hepatic dysfunction, vitamin D status, and glycemic control in children and adolescents with vitamin D deficiency and either type 1 and type 2 diabetes mellitus. PLoS ONE 2014, 9, e99646. [Google Scholar] [CrossRef] [PubMed]
- Minambres, I.; Sanchez-Quesada, J.L.; Vinagre, I.; Sanchez-Hernandez, J.; Urgell, E.; de Leia, A.; Perez, A. Hypovitaminosis D in type 2 diabetes: Relation with features of the metabolic syndrome and glycemic control. Endocr. Res. 2015, 40, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Enciso, P.L.; Wang, L.; Kawahara, Y.; Sakamoto, S.; Shimada, S.; Takeichi, Y.; Takayanagi, R.; Nomura, M. Dietary virtamin D3 improves postprandial hyperglycemia in aged mice. Biochem. Biophys. Res. Commun. 2015, 461, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Leung, P.S. Physiology of the pancreas. Adv. Exp. Med. Biol. 2010, 690, 13–27. [Google Scholar] [PubMed]
- Leung, P.S. Current research of the RAS in T2DM. Adv. Exp. Med. Biol. 2010, 690, 131–153. [Google Scholar] [PubMed]
- Mathieu, C. Vitamin D and diabetes: Where do we stand? Diabetes Res. Clin. Pract. 2015, 108, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Mezza, T.; Muscogiuri, G.; Sorice, G.P.; Prioletta, A.; Salomone, E.; Pontecorvi, A.; Giaccari, A. Vitamin D deficiency: A new risk factor for type 2 diabetes? Ann. Nutr. Metab. 2012, 61, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Boucher, B.J.; Mannan, N.; Noonan, K.; Hales, C.N.; Evans, S.J.W. Glucose intolerance and impairment of insulin secretion in relation to vitamin D deficiency in East London Asians. Diabetologia 1995, 38, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Pajvani, U.B.; Accili, D. The new biology of diabetes. Diabetologia 2015, 58, 2459–2468. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.P.; Chan, L.K.Y.; Leung, P.S. Involvement of the niacin receptor GPR109a in the local control of glucose uptake in small intestine of type 2 diabetic mice. Nutrients 2015, 7, 7543–7561. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; So, W.Y.; Li, S.Y.T.; Cheng, Q.; Boucher, B.J.; Leung, P.S. Niacin-induced hyperglycemia is partially mediated via niacin receptor GPR109a in pancreatic islets. Mol. Cell. Endocriol. 2015, 404, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Sunlight, UV-radiation, vitamin D and skin cancer. Adv. Exp. Med. Biol. 2008, 624, 1–15. [Google Scholar] [PubMed]
- Peechakara, S.V.; Pittas, A.G. Vitamin D as a potential modifier of diabetes risks. Nat. Clin. Pract. Endocrinol. Metab. 2008, 4, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C.; Gysemans, C.; Bouillon, R. Vitamin D and diabetes. Diabetologia 2005, 48, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Ramana, C.V.; Busso, A.S. Stat1-vitamin D receptor interactions antagonize 1,25-dihyroxyvitamin D transcriptional activity and enhance stat1-mediated transcription. Mol. Cell. Biol. 2002, 22, 2777–2787. [Google Scholar] [CrossRef] [PubMed]
- Maestro, B.; Davila, N.; Carranza, M.C.; Calle, C. Identification of a vitamin D response element in the human insulin receptor gene promoter. J. Steroid Biochem. Mol. Biol. 2003, 84, 223–230. [Google Scholar] [CrossRef]
- Eerligh, P.; Koeleman, B.P.; Dudbridge, F.; Bruining, J.G.; Roep, B.O.; Giphart, M.J. Functional genetic polymorphisms in cytokines and metabolic genes as additional genetic markers for susceptibility to develop type 1 diabetes. Genes Immun. 2004, 5, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Leung, P.S.; Cheng, Q. The novel roles of glucagon-like peptide-1, angiotensin II, and vitamin D in islet function. Adv. Exp. Med. Biol. 2010, 654, 339–361. [Google Scholar] [PubMed]
- Alvarez, J.A.; Ashraf, A. Role of vitamin D in insulin secretion and insulin sensitivity for glucose homeostasis. Int. J. Endocrinol. 2010, 2010, 351385. [Google Scholar] [CrossRef] [PubMed]
- Kayaniyil, S.; Vieth, R.; Retnakaran, R.; Knight, J.A.; Qi, Y.; Gerstein, H.C.; Perkins, B.A.; Harris, S.B.; Zinman, B.; Hanley, A.J. Association of vitamin D with insulin resistance and beta-cell dysfunction in subjects at risk for type 2 diabetes. Diabetes Care 2010, 33, 1379–1381. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Sharp, S.J.; Burgess, S.; Scott, R.A.; Imamura, F.; InterAct Consortium; Langenberg, C.; Wareham, N.J.; Forouhi, N.G. Association between circulating 25-hydroxyvitamin D and incident type 2 diabetes: A mendelian randomization study. Lancet Diabetes Endocrinol. 2015, 3, 35–42. [Google Scholar] [CrossRef]
- Heshmat, R.; Tabatabaei-Malazy, O.; Abbaszadeh-Ahranjiani, S.; Shahbazi, S.; Khooshehchin, G.; Bandarian, F.; Larijani, B. Effect of vitamin D on insulin resistance and anthropometric parameters in type 2 diabetes: A randomized double-blind clinical trial. DARU 2012, 20, 10. [Google Scholar] [CrossRef] [PubMed]
- Ryu, O.H.; Lee, S.; Yu, J.; Choi, M.G.; Yoo, H.J.; Mantero, F. A prospective randomized controlled trial of the effects of vitamin D supplementation on long-term glycemic control in type 2 diabetes mellitus of Korea. Endocr. J. 2014, 61, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Elkassaby, S.; Harrison, L.C.; Mazzitelli, N.; Wentworth, J.M.; Colman, P.G.; Spelman, T.; Fourlanos, S. A randomized controlled trial of high dose vitamin D in recent-onset type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 106, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Krul-Poel, Y.H.; Westra, S.; ten Boekel, E.; ter Wee, M.M.; van Schoor, N.M.; van Wijland, H.; Stam, F.; Lips, P.T.; Simsek, S. Effect of vitamin D supplementation on glycemic control in patients with type 2 diabetes (SUNNY Trial): A randomized placebo-controlled trial. Diabetes Care 2015, 38, 1420–1426. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Nolan, C.J. Islet beta cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812. [Google Scholar] [CrossRef] [PubMed]
- Kumashiro, N.; Erion, D.M.; Zhang, D.; Kahn, M.; Beddow, S.A.; Chu, X.; Still, C.D.; Gerhard, G.S.; Han, X.; Dziura, J.; et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2011, 108, 16381–16385. [Google Scholar] [CrossRef] [PubMed]
- Flamment, M.; Hajduch, E.; Ferré, P.; Foufelle, F. New insights into ER stress-induced insulin resistance. Trends Endocrinol. Metab. 2012, 23, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Del Prato, S. Role of glucotoxicity and lipotoxicity in the pathophysiology of Type 2 diabetes mellitus and emerging treatment strategies. Diabet. Med. 2009, 26, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Yabaluri, N.; Bashyam, M.D. Hormonal regulation of gluconeogenic gene transcription in the liver. J. Biosci. 2010, 35, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Roach, P.J.; Cao, Y.; Corbett, C.A.; DePaoli-Roach, A.A.; Farkas, I.; Fiol, C.J.; Flotow, H.; Graves, P.R.; Hardy, T.A.; Hrubey, T.W.; et al. Glycogen metabolism and signal transduction in mammals and yeast. Adv. Enzyme Regul. 1991, 31, 101–120. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose ultilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Carling, D.; Sanders, M.J.; Woods, A. The regulation of AMP-activated protein kinase by upstream kinases. Int. J. Obes. (Lond.) 2008, 32, S55–S59. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.C.; Zierath, J.R. AMP-activated protein kinase signaling in metabolic regulation. J. Clin. Investig. 2006, 116, 1776–1783. [Google Scholar] [CrossRef] [PubMed]
- Barthel, A.; Schmoll, D.; Krüger, K.D.; Roth, R.A.; Joost, H.G. Regulation of the forkhead transcription factor FKHR (FOXO1a) by glucose starvation and AICAR, an activator of AMP-activated protein kinase. Endocrinology 2002, 143, 3183–3186. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.; Giles, A.; Nakamura, K.; Lee, J.W.; Hou, X.; Donmez, G.; Li, J.; Luo, Z.; Walsh, K.; et al. Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB. J. 2011, 25, 1664–1679. [Google Scholar] [CrossRef] [PubMed]
- Kamagate, A.; Kim, D.H.; Zhang, T.; Slusher, S.; Gramignoli, R.; Strom, S.C.; Bertera, S.; Ringquist, S.; Dong, H.H. FoxO1 links hepatic insulin action to endoplasmic reticulum stress. Endocrinology 2010, 151, 3521–3535. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Angelico, F.; Del Ben, M.; Baroni, M.G.; Pozzilli, P.; Morini, S.; Cavallo, M.G. Strong association between non alcoholic fatty liver disease (NAFLD) and low 25(OH) vitamin D levels in an adult population with normal serum liver enzymes. BMC Med. 2011, 9, 85. [Google Scholar] [CrossRef] [PubMed]
- Kwok, R.M.; Torres, D.M.; Harrison, S.A. Vitamin D and nonalcoholic fatty liver disease (NAFLD): Is it more than just an association? Hepatology 2013, 58, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Farese, R.V.; Zechner, R.; Newgard, C.B.; Walther, T.C. The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab. 2012, 15, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Nagle, C.A.; Klett, E.L.; Coleman, R.A. Hepatic triacylglycerol accumulation and insulin resistance. J. Lipid Res. 2009, 50, S74–S79. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Petersen, K.F.; Shulman, G.I. Lipid-induced insulin resistance: Unravelling the mechanism. Lancet 2010, 375, 2267–2277. [Google Scholar] [CrossRef]
- Summers, S.A. Sphingolipids and insulin resistance: The five Ws. Curr. Opin. Lipidol. 2010, 21, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Kumashiro, N.; Beddow, S.A.; Vatner, D.F.; Majumdar, S.K.; Cantley, J.L.; Guebre-Egziabher, F.; Fat, I.; Guigni, B.; Jurczak, M.J.; Birkenfeld, A.L.; et al. Targeting pyruvate carboxylase reduces gluconeogenesis and adiposity and improves insulin resistance. Diabetes 2013, 62, 2183–2194. [Google Scholar] [CrossRef] [PubMed]
- Bell, G.I.; Polonsky, K.S. Diabetes mellitus and genetically programmed defects in beta-cell function. Nature 2001, 414, 788–791. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E. The genetic basis of type 2 diabetes mellitus: Impaired insulin secretion versus impaired insulin sensitivity. Endocr. Rev. 1998, 19, 491–503. [Google Scholar] [PubMed]
- Groop, L. Pathogenesis of type 2 diabetes: The relative contribution of insulin resistance and impaired insulin secretion. Int. J. Clin. Pract. Suppl. 2000, 113, 3–13. [Google Scholar] [PubMed]
- Tuttle, R.; Gill, N.S.; Pugh, W.; Lee, J.P.; Koeberlein, B.; Furth, E.E.; Polonsky, K.S.; Naji, A.; Birnbaum, M.J. Regulation of pancreatic β-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat. Med. 2001, 7, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Mizrachi, E.; Fatrai, S.; Johnson, J.D.; Ohsugi, M.; Otani, K.; Han, Z.; Polonsky, K.S.; Permutt, M.A. Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet β cells. J. Clin. Investig. 2004, 114, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Wrede, C.; Dickson, L.M.; Lingohr, M.K.; Briaud, I.; Rhodes, C.J. Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic β cells (INS-1). J. Biol. Chem. 2002, 277, 49676–49684. [Google Scholar] [CrossRef] [PubMed]
- Jetton, T.L.; Lausier, J.; LaRock, K.; Trotman, W.E.; Larmie, B.; Habibovic, A.; Peshavaria, M.; Leahy, J.L. Mechanisms of compensatory β-cell growth in insulin-resistant rats: Roles of Akt kinase. Diabetes 2005, 54, 2294–2304. [Google Scholar] [CrossRef] [PubMed]
- Martinez, S.; Tanabe, K.; Cras-Méneur, C.; Abumrad, N.A.; Bernal-Mizrachi, E.; Permutt, M.A. Inhibition of Foxo1 protects pancreatic islet beta-cells against fatty acid and endoplasmic reticulum stress-induced apoptosis. Diabetes 2008, 57, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Mussmann, R.; Geese, M.; Harder, F.; Kegel, S.; Andag, U.; Lomow, A.; Burk, U.; Onichtchouk, D.; Dohrmann, C.; Austen, M. Inhibition of GSK3 promotes replication and survival of pancreatic beta cells. J. Biol. Chem. 2007, 282, 12030–12037. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Ohsugi, M.; Liu, Z.G.; Fatrai, S.; Mizrachi, E.B.; Permutt, M.A. Endoplasmic reticulum stress-induced apoptosis is partly mediated by reduced insulin signaling through phosphatidylinositol 3-kinase/Akt and increased glycogen synthase kinase-3β in mouse insulinoma cells. Diabetes 2005, 54, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, E.J.; Prasannarong, M. The role of the renin-angiotensin system in the development of insulin resistance in skeletal muscle. Mol. Cell Endocrinol. 2013, 378, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Lu, C.L.; Wang, Y.; Li, Y.; Li, X.Y. Ang (1–7) protects islet endothelial cells from palmitate-induced apopotosis by AKT, eNOS, p38 MAPK, and JNK pathways. J. Diabetes Res. 2014, 2014, 391476. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Mizuta, M.; Saitoh, Y.; Noma, K.; Ueno, H.; Nakazato, M. Glucagon-like peptide-1 and candesartan additively improve glucolipotoxicity in pancreatic beta-cells. Metabolism 2011, 60, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Leung, P.S. The renin-angiotensin system: Current research progress in the pancreas. In Advances in Experimental Medicine and Biology; Springer: Dordrecht, The Netherlands, 2010; Volume 690, p. 207. [Google Scholar]
- Lau, T.; Carlsson, P.O.; Leung, P.S. Evidence for a local angiotensin-generating system and dose-dependent inhibition of glucose-stimulated insulin release by angiotensin II in isolated pancreatic islets. Diabetologia 2004, 47, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Chu, K.Y.; Lau, T.; Carlsson, P.O.; Leung, P.S. Angiotensin II type 1 receptor blockade improves beta-cell function and glucose tolerance in a mouse model of type 2 diabetes. Diabetes 2006, 55, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Kampf, C.; Lau, T.; Olsson, R.; Leung, P.S.; Carlsson, P.O. Angiotensin II type 1 receptor inhibition improves the blood perfusion, oxygen tension and first phase of glucose-stimulated insulin secretion in revascularized syngeneic mouse islet grafts. Diabetologia 2005, 48, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Chu, K.Y.; Leung, P.S. Angiotensin II Type 1 receptor antagonism mediates uncoupling protein 2-driven oxidative stress and ameliorates pancreatic islet beta-cell function in young Type 2 diabetic mice. Antioxid. Redox Signal. 2007, 9, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Law, P.K.; de Gasparo, M.; Leung, P.S. Combination of the dipeptidyl peptidase IV inhibitor LAF237 with the angiotensin II type 1 receptor antagonist valsartan enhances pancreatic islet morphology and function in a mouse model of type 2 diabetes. J. Pharmacol. Exp. Ther. 2008, 327, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Kong, J.; Wei, M.; Chen, Z.F.; Liu, S.Q.; Cao, L.P. 1,25-Dihydroxyvitamin D3 is a negiatve endocrine regulator of the renin-angiotensin system. J. Clin. Investig. 2002, 110, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Qiao, G.; Uskokovic, M.; Xiang, W.; Zheng, W.; Kong, J. Vitamin D: A negative endocrine regulator of the renin-angiotensin system and blood pressure. J. Steroid Biochem. Mol. Biol. 2004, 90, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Pittas, A.G.; Lau, J.; Hu, F.B.; Dawson-Gughes, B. The role of vitamin D and calcium in type 2 diabetes: A systemic review and meta-analysis. J. Clin. Endocrinol. Metab. 2007, 92, 2017–2029. [Google Scholar] [CrossRef] [PubMed]
- Pittas, A.G.; Dawson-Gughes, B.; Li, T.; Van Dam, R.M.; Willett, W.C.; Manson, J.E.; Hu, F.B. Vitamin D and calcium intake in relation to type 2 diabetes in women. Diabetes Care 2006, 29, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Leung, P.S. An update on the islet renin-angiotensin system. Peptides 2011, 32, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Leung, P.S.; Boucher, B.J. The roles of vitamin D in modulation of the pancreatic renin-angiotensin system. In Angiotensin Research Progress; Miura, H., Sasaki, Y., Eds.; Nova Science Publishers: New York, NY, USA, 2008; pp. 201–220. [Google Scholar]
- Cheng, Q.; Li, Y.C.; Boucher, B.J.; Leung, P.S. A novel role for vitamin D: Modulation of expression and function of the local renin-angiotensin system in mouse pancreatic islets. Diabetologia 2011, 54, 2077–2081. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Boucher, B.J.; Leung, P.S. Modulation of hypovitaminosis D-induced islet dysfunction and insulin resistance through direct suppression of the pancreatic islet renin-angiotensin system in mice. Diabetologia 2013, 56, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Leung, P.S.; Ng, K.Y. Current progress in stem cell research and its potential for islet cell transplantation. Curr. Mol. Med. 2013, 13, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Suen, P.M.; Chan, J.C.; Lau, T.K.; Yao, K.M.; Leung, P.S. PDZ-domain-containing 2 (PDZD2) is a novel factor that affects the growth and differentiation of human fetal pancreatic progenitor cells. Int. J. Biochem. Cell Biol. 2008, 40, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.K.; Suen, P.M.; Lau, T.K.; Ko, W.H.; Yao, K.M.; Leung, P.S. PDZ-domain containing-2 (PDZD2) drives the maturity of human fetal pancreatic progenitor-derived islet-like cell clusters with functional responsiveness against membrane depolarization. Stem Cell Dev. 2009, 18, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.K.; Liang, J.; Ma, M.T.; Leung, P.S. Angiotensin II type 2 receptor is critical for the development of human fetal pancreatic progenitor cells into islet-like cell clusters and their potential for transplantation. Stem Cells 2012, 30, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.Y.; Ma, M.T.; Leung, K.K.; Leung, P.S. Vitamin D and vitamin A receptor expression and the proliferative effects of ligand activation of these receptors on the development of pancreatic progenitor cells derived from human fetal pancreas. Stem Cell Rev. 2011, 7, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Niederreither, K.; Dollé, P. Retinoic acid in development: Towards an integrated view. Nat. Rev. Genet. 2008, 9, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Oström, M.; Loffler, K.A.; Edfalk, S. Retinoic acid promotes the generation of pancreatic endocrine progenitor cells and their further differentiation into beta-cells. PLoS ONE 2008, 3, e2841. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Hou, L.; Tang, F. Inducing embryonic stem cells to differentiate into pancreatic beta cells by a novel three-step approach with activin A and all-trans retinoic acid. Stem Cells 2005, 23, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Ng, K.Y.; Cheng, Q.; Xia, Y.; Wang, C.C.; Leung, P.S. Human fetal liver stromal cells co-culture enhances the differentiation of pancreatic progenitor cells into islet-like cell clusters. Stem Cell Rev. 2014, 10, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Seet, E.L.; Yee, J.K.; Jellyman, J.K.; Hun, G.; Ross, M.G. Maternal high-fat-diet programs rat offspring liver fatty acid metabolism. Lipids 2015, 50, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.R.; Lin, L.Q.; Ma, H.; Li, Y.; Sun, C.H. Association between vitamin D receptor gene polymorphism (TaqI) and obesity in Chinese population. J. Genet. 2015, 94, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Wortsman, J.; Matsuoka, L.Y.; Chen, T.C.; Lu, Z.; Holick, M.F. Decreased bioavailability of vitamin D in obesity. Am. J. Clin. Nutr. 2000, 72, 690–693. [Google Scholar] [PubMed]
- Boucher, B.J.; Leung, P.S. “Maternal high-fat-diet programs rat offspring liver fatty acid metabolism”: Might reduced vitamin D availability due to increases in maternal body fat contribute to this effect? Lipids 2015, 50, 837–838. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leung, P.S. The Potential Protective Action of Vitamin D in Hepatic Insulin Resistance and Pancreatic Islet Dysfunction in Type 2 Diabetes Mellitus. Nutrients 2016, 8, 147. https://doi.org/10.3390/nu8030147
Leung PS. The Potential Protective Action of Vitamin D in Hepatic Insulin Resistance and Pancreatic Islet Dysfunction in Type 2 Diabetes Mellitus. Nutrients. 2016; 8(3):147. https://doi.org/10.3390/nu8030147
Chicago/Turabian StyleLeung, Po Sing. 2016. "The Potential Protective Action of Vitamin D in Hepatic Insulin Resistance and Pancreatic Islet Dysfunction in Type 2 Diabetes Mellitus" Nutrients 8, no. 3: 147. https://doi.org/10.3390/nu8030147
APA StyleLeung, P. S. (2016). The Potential Protective Action of Vitamin D in Hepatic Insulin Resistance and Pancreatic Islet Dysfunction in Type 2 Diabetes Mellitus. Nutrients, 8(3), 147. https://doi.org/10.3390/nu8030147