Hepatic Retinyl Ester Hydrolases and the Mobilization of Retinyl Ester Stores



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Brief Overview of the Role of Different Liver Cell Types in Hepatic Vitamin A Turnover
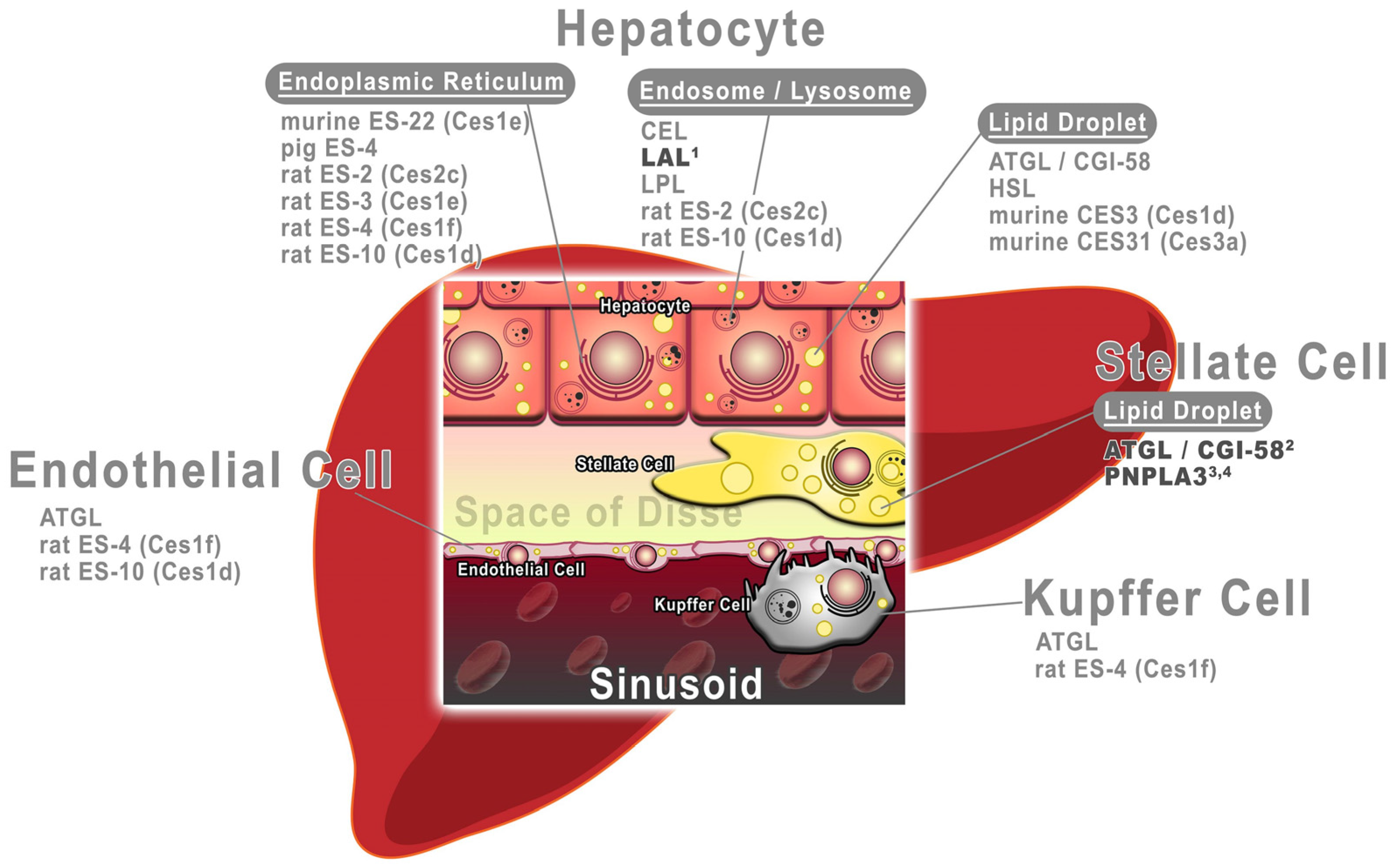
2.1. Parenchymal Cells in Vitamin A Turnover
2.2. Non-Parenchymal Stellate Cells in Vitamin A Turnover
2.3. Other Non-Parenchymal Cell Types
3. Hepatic Retinyl Ester (RE) Hydrolases
3.1. Retinyl Ester Hydrolases of Hepatocytes
3.1.1. Retinyl Ester Hydrolases in the Endocytic System of Hepatocytes
3.1.2. Retinyl Ester Hydrolases of the Endoplasmic Reticulum of Hepatocytes
3.1.3. Retinyl Ester Hydrolases of the Lipid Droplet of Hepatocytes
3.2. RE Hydrolases of Hepatic Stellate Cells
3.3. RE Hydrolases of Other Non-Parenchymal Cells
4. Pathophysiological Processes Associated with Mobilization of Hepatic RE Stores
4.1. RE Mobilization upon Alcohol-Related Liver Diseases/Alcoholic Liver Cirrhosis
4.2. RE Mobilization upon Nonalcoholic Fatty Liver Disease
4.3. RE Mobilization upon Viral Hepatitis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- O’Byrne, S.M.; Blaner, W.S. Retinol and retinyl esters: Biochemistry and physiology. J. Lipid Res. 2013, 54, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Reboul, E. Absorption of vitamin A and carotenoids by the enterocyte: Focus on transport proteins. Nutrients 2013, 5, 3563–3581. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.C.; Zolfaghari, R. Functions and Actions of Retinoids and Carotenoids: Building on the Vision of James Allen Olson Regulation of Hepatic Retinol Metabolism: Perspectives from Studies on Vitamin A Status. J. Nutr. 2004, 134, 269S–275S. [Google Scholar] [PubMed]
- Huang, P.; Chandra, V.; Rastinejad, F. Retinoic acid actions through mammalian nuclear receptors. Chem. Rev. 2014, 114, 233–254. [Google Scholar] [CrossRef] [PubMed]
- Quadro, L.; Blaner, W.S.; Hamberger, L.; van Gelder, R.N.; Vogel, S.; Piantedosi, R.; Gouras, P.; Colantuoni, V.; Gottesman, M.E. Muscle expression of human retinol-binding protein (RBP). Suppression of the visual defect of RBP knockout mice. J. Biol. Chem. 2002, 277, 30191–30197. [Google Scholar] [CrossRef] [PubMed]
- Blaner, W.S.; Li, Y.; Brun, P.-J.; Yuen, J.J.; Lee, S.-A.; Clugston, R.D. Vitamin A Absorption, Storage and Mobilization. Subcell. Biochem. 2016, 81, 95–125. [Google Scholar] [PubMed]
- Shirakami, Y.; Lee, S.-A.; Clugston, R.D.; Blaner, W.S. Hepatic metabolism of retinoids and disease associations. Biochim. Biophys. Acta 2012, 1821, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Quadro, L.; Gamble, M.V.; Vogel, S.; Lima, A.A.; Piantedosi, R.; Moore, S.R.; Colantuoni, V.; Gottesman, M.E.; Guerrant, R.L.; Blaner, W.S. Retinol and retinol-binding protein: Gut integrity and circulating immunoglobulins. J. Infect. Dis. 2000, S97–S102. [Google Scholar] [CrossRef] [PubMed]
- Green, M.H.; Green, J.B.; Berg, T.; Norum, K.R.; Blomhoff, R. Changes in hepatic parenchymal and nonparenchymal cell vitamin A content during vitamin A depletion in the rat. J. Nutr. 1988, 118, 1331–1335. [Google Scholar] [PubMed]
- Tanumihardjo, S.A. Vitamin A: Biomarkers of nutrition for development. Am. J. Clin. Nutr. 2011, 94, 658S–665S. [Google Scholar] [CrossRef] [PubMed]
- Senoo, H.; Yoshikawa, K.; Morii, M.; Miura, M.; Imai, K.; Mezaki, Y. Hepatic stellate cell (vitamin A-storing cell) and its relative—Past, present and future Discovery and re-discovery of HSCs. Cell Biol. Int. 2010, 34, 1247–1272. [Google Scholar] [CrossRef] [PubMed]
- Chen, G. The link between Hepatic Vitamin A Metabolism and Nonalcoholic Fatty Liver Disease. Curr. Drug Targets 2015, 16, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Puche, J.E.; Saiman, Y.; Friedman, S.L. Hepatic stellate cells and liver fibrosis. Compr. Physiol. 2013, 3, 1473–1492. [Google Scholar] [PubMed]
- D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A metabolism: An update. Nutrients 2011, 3, 63–103. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, H.; Nakamura, M.; Komori, A.; Migita, K.; Shimoda, S. Liver architecture, cell function, and disease. Semin. Immunopathol. 2009, 31, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Blouin, A.; Bolender, R.P.; Weibel, E.R. Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study. J. Cell Biol. 1977, 72, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Blaner, W.S.; Hendriks, H.F.; Brouwer, A.; de Leeuw, A.M.; Knook, D.L.; Goodman, D.S. Retinoids, retinoid-binding proteins, and retinyl palmitate hydrolase distributions in different types of rat liver cells. J. Lipid Res. 1985, 26, 1241–1251. [Google Scholar] [PubMed]
- Yu, K.C.; Cooper, A.D. Postprandial lipoproteins and atherosclerosis. Front. Biosci. 2001, 6, D332–D354. [Google Scholar] [CrossRef] [PubMed]
- Groot, P.H.; van Berkel, T.J.; van Tol, A. Relative contributions of parenchymal and non-parenchymal (sinusoidal) liver cells in the uptake of chylomicron remnants. Metabolism 1981, 30, 792–797. [Google Scholar] [CrossRef]
- Hussain, M.M. A proposed model for the assembly of chylomicrons. Atherosclerosis 2000, 148, 1–15. [Google Scholar] [CrossRef]
- Santamarina-Fojo, S.; Haudenschild, C. Role of hepatic and lipoprotein lipase in lipoprotein metabolism and atherosclerosis: Studies in transgenic and knockout animal models and somatic gene transfer. Int. J. Tissue React. 2000, 22, 39–47. [Google Scholar] [PubMed]
- Braet, F.; Wisse, E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: A review. Comp. Hepatol. 2002, 1, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjøen, T.; Bjerkelund, T.; Blomhoff, H.K.; Norum, K.R.; Berg, T.; Blomhoff, R. Liver takes up retinol-binding protein from plasma. J. Biol. Chem. 1987, 262, 10926–10930. [Google Scholar] [PubMed]
- Smeland, S.; Bjerknes, T.; Malaba, L.; Eskild, W.; Norum, K.R.; Blomhoff, R. Tissue distribution of the receptor for plasma retinol-binding protein. Biochem. J. 1995, 305, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Alapatt, P.; Guo, F.; Komanetsky, S.M.; Wang, S.; Cai, J.; Sargsyan, A.; Rodríguez Díaz, E.; Bacon, B.T.; Aryal, P.; Graham, T.E. Liver retinol transporter and receptor for serum retinol-binding protein (RBP4). J. Biol. Chem. 2013, 288, 1250–1265. [Google Scholar] [CrossRef] [PubMed]
- Bouillet, P.; Sapin, V.; Chazaud, C.; Messaddeq, N.; Décimo, D.; Dollé, P.; Chambon, P. Developmental expression pattern of Stra6, a retinoic acid-responsive gene encoding a new type of membrane protein. Mech. Dev. 1997, 63, 173–186. [Google Scholar] [CrossRef]
- Quadro, L.; Blaner, W.S.; Hamberger, L.; Novikoff, P.M.; Vogel, S.; Piantedosi, R.; Gottesman, M.E.; Colantuoni, V. The role of extrahepatic retinol binding protein in the mobilization of retinoid stores. J. Lipid Res. 2004, 45, 1975–1982. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.L.; Goodman, D.S. Studies on the metabolism of retinol-binding protein by primary hepatocytes from retinol-deficient rats. J. Cell. Physiol. 1987, 130, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wongsiriroj, N.; Blaner, W.S. The multifaceted nature of retinoid transport and metabolism. Hepatobiliary Surg. Nutr. 2014, 3, 126–139. [Google Scholar] [PubMed]
- Harrison, E.H.; Gad, M.Z.; Ross, A.C. Hepatic uptake and metabolism of chylomicron retinyl esters: Probable role of plasma membrane/endosomal retinyl ester hydrolases. J. Lipid Res. 1995, 36, 1498–1506. [Google Scholar] [PubMed]
- Napoli, J.L. Functions of Intracellular Retinoid Binding-Proteins. Subcell. Biochem. 2016, 81, 21–76. [Google Scholar] [PubMed]
- Herr, F.M.; Ong, D.E. Differential interaction of lecithin-retinol acyltransferase with cellular retinol binding proteins. Biochemistry 1992, 31, 6748–6755. [Google Scholar] [CrossRef] [PubMed]
- Boerman, M.H.; Napoli, J.L. Cholate-independent retinyl ester hydrolysis. Stimulation by Apo-cellular retinol-binding protein. J. Biol. Chem. 1991, 266, 22273–22278. [Google Scholar] [PubMed]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Blaner, W.S.; Dixon, J.L.; Moriwaki, H.; Martino, R.A.; Stein, O.; Stein, Y.; Goodman, D.S. Studies on the in vivo transfer of retinoids from parenchymal to stellate cells in rat liver. Eur. J. Biochem. 1987, 164, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Quiroga, A.D.; Lehner, R. Analysis of lipid droplets in hepatocytes. Methods Cell Biol. 2013, 116, 107–127. [Google Scholar] [PubMed]
- Quiroga, A.D.; Lehner, R. Role of endoplasmic reticulum neutral lipid hydrolases. Trends Endocrinol. Metab. 2011, 22, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gilham, D.; Lehner, R. Proteomic and lipid characterization of apolipoprotein B-free luminal lipid droplets from mouse liver microsomes: Implications for very low density lipoprotein assembly. J. Biol. Chem. 2007, 282, 33218–33226. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Evason, K.J.; Asahina, K.; Stainier, D.Y.R. Hepatic stellate cells in liver development, regeneration, and cancer. J. Clin. Investig. 2013, 123, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.C.; Piantedosi, R.; Libien, J. Hepatic stellate cell lipid droplets: A specialized lipid droplet for retinoid storage. Biochim. Biophys. Acta 2009, 1791, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, V.A.; Martucci, R.B.; Trugo, L.C.; Borojevic, R. Hepatic stellate cells uptake of retinol associated with retinol-binding protein or with bovine serum albumin. J. Cell. Biochem. 2003, 90, 792–805. [Google Scholar] [CrossRef] [PubMed]
- Sauvant, P.; Sapin, V.; Alexandre-Gouabau, M.C.; Dodeman, I.; Delpal, S.; Quadro, L.; Partier, A.; Abergel, A.; Colantuoni, V.; Rock, E.; et al. Retinol mobilization from cultured rat hepatic stellate cells does not require retinol binding protein synthesis and secretion. Int. J. Biochem. Cell Biol. 2001, 33, 1000–1012. [Google Scholar] [CrossRef]
- Andersen, K.B.; Nilsson, A.; Blomhoff, H.K.; Oyen, T.B.; Gabrielsen, O.S.; Norum, K.R.; Blomhoff, R. Direct mobilization of retinol from hepatic perisinusoidal stellate cells to plasma. J. Biol. Chem. 1992, 267, 1340–1344. [Google Scholar] [PubMed]
- Li, J.-T.; Liao, Z.-X.; Ping, J.; Xu, D.; Wang, H. Molecular mechanism of hepatic stellate cell activation and antifibrotic therapeutic strategies. J. Gastroenterol. 2008, 43, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Trautwein, C. Mechanisms of liver fibrosis resolution. J. Hepatol. 2015, 63, 1038–1039. [Google Scholar] [CrossRef] [PubMed]
- Wake, K. Karl Wilhelm Kupffer And His Contributions To Modern Hepatology. Comp. Hepatol. 2004, 3, S2. [Google Scholar] [CrossRef] [PubMed]
- Bilzer, M.; Roggel, F.; Gerbes, A.L. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006, 26, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.H.; Kitamura, T. The vitamin A content and retinol esterifying activity of a Kupffer cell fraction of liver. J. Histochem. Cytochem. 1972, 20, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Dijk, M.C.M.; Ziere, G.J.; Berkel, T.J.C. Characterization of the chylomicron-remnant-recognition sites on parenchymal and Kupffer cells of rat liver Selective inhibition of parenchymal cell recognition by lactoferrin. Eur. J. Biochem. 1992, 205, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Lippiello, P.M.; Sisson, P.J.; Waite, M. The uptake and metabolism of chylomicron-remnant lipids by rat liver parenchymal and non-parenchymal cells in vitro. Biochem. J. 1985, 232, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Heur, M.; Duanmu, M.; Grabowski, G.A.; Hui, D.Y.; Witte, D.P.; Mishra, J. Lysosomal acid lipase-deficient mice: Depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. J. Lipid Res. 2001, 42, 489–500. [Google Scholar] [PubMed]
- Sørensen, K.K.; Simon-Santamaria, J.; McCuskey, R.S.; Smedsrød, B. Liver Sinusoidal Endothelial Cells. Compr. Physiol. 2015, 5, 1751–1774. [Google Scholar] [PubMed]
- Green, M.H.; Green, J.B.; Berg, T.; Norum, K.R.; Blomhoff, R. Vitamin A metabolism in rat liver: A kinetic model. Am. J. Physiol. 1993, 264, G509–G521. [Google Scholar] [PubMed]
- Green, M.H.; Uhl, L.; Green, J.B. A multicompartmental model of vitamin A kinetics in rats with marginal liver vitamin A stores. J. Lipid Res. 1985, 26, 806–818. [Google Scholar] [PubMed]
- Hendriks, H.F.; Blaner, W.S.; Wennekers, H.M.; Piantedosi, R.; Brouwer, A.; de Leeuw, A.M.; Goodman, D.S.; Knook, D.L. Distributions of retinoids, retinoid-binding proteins and related parameters in different types of liver cells isolated from young and old rats. Eur. J. Biochem. 1988, 171, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Garrity, A.G.; Xu, H. Regulation of membrane trafficking by signalling on endosomal and lysosomal membranes. J. Physiol. 2013, 591, 4389–4401. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Taschler, U.; Preiss-Landl, K.; Wongsiriroj, N.; Zimmermann, R.; Lass, A. Retinyl ester hydrolases and their roles in vitamin A homeostasis. Biochim. Biophys. Acta 2012, 1821, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Vergés, M.; Bensadoun, A.; Herz, J.; Belcher, J.D.; Havel, R.J. Endocytosis of hepatic lipase and lipoprotein lipase into rat liver hepatocytes in vivo is mediated by the low density lipoprotein receptor-related protein. J. Biol. Chem. 2004, 279, 9030–9036. [Google Scholar] [CrossRef] [PubMed]
- Blaner, W.S.; Obunike, J.C.; Kurlandsky, S.B.; al-Haideri, M.; Piantedosi, R.; Deckelbaum, R.J.; Goldberg, I.J. Lipoprotein lipase hydrolysis of retinyl ester. Possible implications for retinoid uptake by cells. J. Biol. Chem. 1994, 269, 16559–16565. [Google Scholar] [PubMed]
- Harrison, E.H. Lipases and carboxylesterases: Possible roles in the hepatic utilization of vitamin A. J. Nutr. 2000, 130, 340S–344S. [Google Scholar] [PubMed]
- Sun, G.; Alexson, S.E.; Harrison, E.H. Purification and characterization of a neutral, bile salt-independent retinyl ester hydrolase from rat liver microsomes. Relationship To rat carboxylesterase ES-2. J. Biol. Chem. 1997, 272, 24488–24493. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Yang, D.; Bullock, P.; Parkinson, A. Rat serum carboxylesterase. Cloning, expression, regulation, and evidence of secretion from liver. J. Biol. Chem. 1995, 270, 19128–19134. [Google Scholar] [PubMed]
- Medda, S.; Proia, R.L. The carboxylesterase family exhibits C-terminal sequence diversity reflecting the presence or absence of endoplasmic-reticulum-retention sequences. Eur. J. Biochem. 1992, 206, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Harrison, E.H.; Fisher, E.A. Molecular cloning of the cDNA for rat hepatic, bile salt-dependent cholesteryl ester/retinyl ester hydrolase demonstrates identity with pancreatic carboxylester lipase. Exp. Biol. Med. 1997, 215, 186–191. [Google Scholar] [CrossRef]
- Harrison, E.H.; Rojas, C.J.; Kempner, E.S. Size of the catalytically active unit of rat hepatic carboxylester lipase in the presence and absence of bile salt. Biochim. Biophys. Acta Lipids Lipid Metab. 1997, 1347, 177–182. [Google Scholar] [CrossRef]
- Winkler, K.E.; Harrison, E.H.; Marsh, J.B.; Glick, J.M.; Ross, A.C. Characterization of a bile salt-dependent cholesteryl ester hydrolase activity secreted from HepG2 cells. Biochim. Biophys. Acta 1992, 1126, 151–158. [Google Scholar] [CrossRef]
- Torsvik, J.; Johansson, B.B.; Dalva, M.; Marie, M.; Fjeld, K.; Johansson, S.; Bjørkøy, G.; Saraste, J.; Njølstad, P.R.; Molven, A. Endocytosis of secreted carboxyl ester lipase in a syndrome of diabetes and pancreatic exocrine dysfunction. J. Biol. Chem. 2014, 289, 29097–29111. [Google Scholar] [CrossRef] [PubMed]
- Van Bennekum, A.M.; Li, L.; Piantedosi, R.; Shamir, R.; Vogel, S.; Fisher, E.A.; Blaner, W.S.; Harrison, E.H. Carboxyl ester lipase overexpression in rat hepatoma cells and CEL deficiency in mice have no impact on hepatic uptake or metabolism of chylomicron-retinyl ester. Biochemistry 1999, 38, 4150–4156. [Google Scholar] [CrossRef] [PubMed]
- Linke, T.; Dawson, H.; Harrison, E.H. Isolation and characterization of a microsomal acid retinyl ester hydrolase. J. Biol. Chem. 2005, 280, 23287–23294. [Google Scholar] [CrossRef] [PubMed]
- Grumet, L.; Eichmann, T.O.; Taschler, U.; Zierler, K.A.; Leopold, C.; Moustafa, T.; Radovic, B.; Romauch, M.; Yan, C.; Du, H.; et al. Lysosomal acid lipase hydrolyzes retinyl ester and affects retinoid turnover. J. Biol. Chem. 2016, 291, 17977–17987. [Google Scholar] [CrossRef] [PubMed]
- Tylki-Szymańska, A.; Jurecka, A. Lysosomal acid lipase deficiency: Wolman disease and cholesteryl ester storage disease. Pril. (Makedon. Akad. Nauk. Umet. Odd. Med. Nauki) 2014, 35, 99–106. [Google Scholar] [PubMed]
- Du, H.; Duanmu, M.; Witte, D.; Grabowski, G.A. Targeted disruption of the mouse lysosomal acid lipase gene: Long-term survival with massive cholesteryl ester and triglyceride storage. Hum. Mol. Genet. 1998, 7, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Mercier, M.; Forget, A.; Grolier, P.; Azais-Braesco, V. Hydrolysis of retinyl esters in rat liver. Description of a lysosomal activity. Biochim. Biophys. Acta Lipids Lipid Metab. 1994, 1212, 176–182. [Google Scholar] [CrossRef]
- Blomhoff, R.; Eskild, W.; Kindberg, G.M.; Prydz, K.; Berg, T. Intracellular transport of endocytosed chylomicron [3H]retinyl ester in rat liver parenchymal cells. Evidence for translocation of a [3H]retinoid from endosomes to endoplasmic reticulum. J. Biol. Chem. 1985, 260, 13566–13570. [Google Scholar] [PubMed]
- Hagen, E.; Myhre, A.M.; Tjelle, T.E.; Berg, T.; Norum, K.R. Retinyl esters are hydrolyzed in early endosomes of J774 macrophages. J. Lipid Res. 1999, 40, 309–317. [Google Scholar] [PubMed]
- Yen, C.-L.E.; Monetti, M.; Burri, B.J.; Farese, R.V. The triacylglycerol synthesis enzyme DGAT1 also catalyzes the synthesis of diacylglycerols, waxes, and retinyl esters. J. Lipid Res. 2005, 46, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Moise, A.R.; Golczak, M.; Imanishi, Y.; Palczewski, K. Topology and membrane association of lecithin: retinol acyltransferase. J. Biol. Chem. 2007, 282, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, S.M.; Wongsiriroj, N.; Libien, J.; Vogel, S.; Goldberg, I.J.; Baehr, W.; Palczewski, K.; Blaner, W.S. Retinoid absorption and storage is impaired in mice lacking lecithin: Retinol acyltransferase (LRAT). J. Biol. Chem. 2005, 280, 35647–35657. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Gudas, L.J. Disruption of the lecithin: Retinol acyltransferase gene makes mice more susceptible to vitamin A deficiency. J. Biol. Chem. 2005, 280, 40226–40234. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.-X.; Diehl, A.M. Diacylglycerol acyltranferase 1 anti-sense oligonucleotides reduce hepatic fibrosis in mice with nonalcoholic steatohepatitis. Hepatology 2008, 47, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Holmes, R.S.; Wright, M.W.; Laulederkind, S.J.F.; Cox, L.A.; Hosokawa, M.; Imai, T.; Ishibashi, S.; Lehner, R.; Miyazaki, M.; Perkins, E.J.; et al. Recommended nomenclature for five mammalian carboxylesterase gene families: Human, mouse, and rat genes and proteins. Mamm. Genome 2010, 21, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, M. Structure and catalytic properties of carboxylesterase isozymes involved in metabolic activation of prodrugs. Molecules 2008, 13, 412–431. [Google Scholar] [CrossRef] [PubMed]
- Mentlein, R.; Heymann, E. Hydrolysis of retinyl esters by non-specific carboxylesterases from rat liver endoplasmic reticulum. Biochem. J. 1987, 245, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Taschler, U.; Wolinski, H.; Seper, A.; Tamegger, S.N.; Graf, M.; Kohlwein, S.D.; Haemmerle, G.; Zimmermann, R.; Zechner, R.; et al. Esterase 22 and beta-glucuronidase hydrolyze retinoids in mouse liver. J. Lipid Res. 2009, 50, 2514–2523. [Google Scholar] [CrossRef] [PubMed]
- Schindler, R.; Mentlein, R.; Feldheim, W. Purification and characterization of retinyl ester hydrolase as a member of the non-specific carboxylesterase supergene family. Eur. J. Biochem. 1998, 251, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Ronne, H.; Ocklind, C.; Wiman, K.; Rask, L.; Obrink, B.; Peterson, P.A. Ligand-dependent regulation of intracellular protein transport: Effect of vitamin A on the secretion of the retinol-binding protein. J. Cell Biol. 1983, 96, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Mello, T.; Nakatsuka, A.; Fears, S.; Davis, W.; Tsukamoto, H.; Bosron, W.F.; Sanghani, S.P.; Vita, A. Expression of carboxylesterase and lipase genes in rat liver cell-types. Biochem. Biophys. Res. Commun. 2008, 374, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Sanghani, S.P.; Davis, W.I.; Dumaual, N.G.; Mahrenholz, A.; Bosron, W.F. Identification of microsomal rat liver carboxylesterases and their activity with retinyl palmitate. Eur. J. Biochem. 2002, 269, 4387–4398. [Google Scholar] [CrossRef] [PubMed]
- Ström, K.; Gundersen, T.E.; Hansson, O.; Lucas, S.; Fernandez, C.; Blomhoff, R.; Holm, C. Hormone-sensitive lipase (HSL) is also a retinyl ester hydrolase: Evidence from mice lacking HSL. FASEB J. 2009, 23, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.N.; Ables, G.P.; Otlivanchik, O.A.; Schoiswohl, G.; Zechner, R.; Blaner, W.S.; Goldberg, I.J.; Schwabe, R.F.; Chua, S.C.; Huang, L.-S.; et al. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J. Biol. Chem. 2008, 283, 13087–13099. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, M.; Osuga, J.-I.; Yahagi, N.; Okazaki, H.; Tamura, Y.; Igarashi, M.; Takase, S.; Harada, K.; Okazaki, S.; Iizuka, Y.; et al. Hormone-sensitive lipase is involved in hepatic cholesteryl ester hydrolysis. J. Lipid Res. 2008, 49, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Doll, J.A.; Gattu, A.K.; Shugrue, C.; Cornwell, M.; Fitchev, P.; Crawford, S.E. Anti-angiogenic pigment epithelium-derived factor regulates hepatocyte triglyceride content through adipose triglyceride lipase (ATGL). J. Hepatol. 2008, 48, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Taschler, U.; Schreiber, R.; Chitraju, C.; Grabner, G.F.; Romauch, M.; Wolinski, H.; Haemmerle, G.; Breinbauer, R.; Zechner, R.; Lass, A.; et al. Adipose triglyceride lipase is involved in the mobilization of triglyceride and retinoid stores of hepatic stellate cells. Biochim. Biophys. Acta 2015, 1851, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Tuohetahuntila, M.; Molenaar, M.R.; Spee, B.; Brouwers, J.F.; Houweling, M.; Vaandrager, A.B.; Helms, J.B. ATGL and DGAT1 are involved in the turnover of newly synthesized triacylglycerols in hepatic stellate cells. J. Lipid Res. 2016, 57, 1162–1174. [Google Scholar] [CrossRef] [PubMed]
- Crunk, A.E.; Monks, J.; Murakami, A.; Jackman, M.; MacLean, P.S.; Ladinsky, M.; Bales, E.S.; Cain, S.; Orlicky, D.J.; McManaman, J.L. Dynamic regulation of hepatic lipid droplet properties by diet. PLoS ONE 2013, 8, e67631. [Google Scholar] [CrossRef] [PubMed]
- Lehner, R.; Cui, Z.; Vance, D.E. Subcellullar localization, developmental expression and characterization of a liver triacylglycerol hydrolase. Biochem. J. 1999, 338, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Gaustad, R.; Berg, T.; Fonnum, F. Heterogeneity of carboxylesterases in rat liver cells. Biochem. Pharmacol. 1992, 44, 827–829. [Google Scholar] [CrossRef]
- Lehner, R.; Vance, D.E. Cloning and expression of a cDNA encoding a hepatic microsomal lipase that mobilizes stored triacylglycerol. Biochem. J. 1999, 343, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wei, E.; Ben Ali, Y.; Lyon, J.; Wang, H.; Nelson, R.; Dolinsky, V.W.; Dyck, J.R.B.; Mitchell, G.; Korbutt, G.S.; Lehner, R. Loss of TGH/Ces3 in mice decreases blood lipids, improves glucose tolerance, and increases energy expenditure. Cell Metab. 2010, 11, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Wei, E.; Wang, S.P.; Quiroga, A.D.; Li, L.; Di Pardo, A.; van der Veen, J.; Sipione, S.; Mitchell, G.A.; Lehner, R. Liver specific inactivation of carboxylesterase 3/triacylglycerol hydrolase decreases blood lipids without causing severe steatosis in mice. Hepatology 2012, 56, 2154–2162. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, V.W.; Gilham, D.; Alam, M.; Vance, D.E.; Lehner, R. Triacylglycerol hydrolase: Role in intracellular lipid metabolism. Cell. Mol. Life Sci. 2004, 61, 1633–1651. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lopez, N.; Singh, R. Autophagy and lipid droplets in the liver. Annu. Rev. Nutr. 2015, 35, 215–237. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Czaja, M.J. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013, 20, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Lass, A.; Zimmermann, R.; Oberer, M.; Zechner, R. Lipolysis—A highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog. Lipid Res. 2011, 50, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Zechner, R.; Kienesberger, P.C.; Haemmerle, G.; Zimmermann, R.; Lass, A. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J. Lipid Res. 2009, 50, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Eichmann, T.O.; Grumet, L.; Taschler, U.; Hartler, J.; Heier, C.; Woblistin, A.; Pajed, L.; Kollroser, M.; Rechberger, G.; Thallinger, G.G.; et al. ATGL and CGI-58 are lipid droplet proteins of the hepatic stellate cell line HSC-T6. J. Lipid Res. 2015, 56, 1972–1984. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Yang, X.; Qing, K.; Ou-Yang, Y. Roles of the lipid metabolism in hepatic stellate cells activation. Chin. Med. Sci. J. 2013, 28, 233–236. [Google Scholar] [CrossRef]
- Testerink, N.; Ajat, M.; Houweling, M.; Brouwers, J.F.; Pully, V.V.; van Manen, H.-J.; Otto, C.; Helms, J.B.; Vaandrager, A.B. Replacement of retinyl esters by polyunsaturated triacylglycerol species in lipid droplets of hepatic stellate cells during activation. PLoS ONE 2012, 7, e34945. [Google Scholar] [CrossRef] [PubMed]
- Vicente, C.P.; Guaragna, R.M.; Borojevic, R. Lipid metabolism during in vitro induction of the lipocyte phenotype in hepatic stellate cells. Mol. Cell. Biochem. 1997, 168, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Roh, C.; Roduit, R.; Thorens, B.; Fried, S.; Kandror, K.V. Lipoprotein lipase and leptin are accumulated in different secretory compartments in rat adipocytes. J. Biol. Chem. 2001, 276, 35990–35994. [Google Scholar] [CrossRef] [PubMed]
- Pirazzi, C.; Valenti, L.; Motta, B.M.; Pingitore, P.; Hedfalk, K.; Mancina, R.M.; Burza, M.A.; Indiveri, C.; Ferro, Y.; Montalcini, T.; et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet. 2014, 23, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, P.; Pirazzi, C.; Mancina, R.M.; Motta, B.M.; Indiveri, C.; Pujia, A.; Montalcini, T.; Hedfalk, K.; Romeo, S. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim. Biophys. Acta 2014, 1841, 574–580. [Google Scholar] [CrossRef] [PubMed]
- He, S.; McPhaul, C.; Li, J.Z.; Garuti, R.; Kinch, L.; Grishin, N.V.; Cohen, J.C.; Hobbs, H.H. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J. Biol. Chem. 2010, 285, 6706–6715. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Schoiswohl, G.; Chitraju, C.; Paar, M.; Cornaciu, I.; Rangrez, A.Y.; Wongsiriroj, N.; Nagy, H.M.; Ivanova, P.T.; Scott, S.A.; et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012, 15, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Al-Serri, A.; Daly, A.K.; Galmozzi, E.; Rametta, R.; Dongiovanni, P.; Nobili, V.; Mozzi, E.; Roviaro, G.; Vanni, E.; et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Basantani, M.K.; Sitnick, M.T.; Cai, L.; Brenner, D.S.; Gardner, N.P.; Li, J.Z.; Schoiswohl, G.; Yang, K.; Kumari, M.; Gross, R.W.; et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J. Lipid Res. 2011, 52, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Ochi, T.; Munekage, K.; Ono, M.; Higuchi, T.; Tsuda, M.; Hayashi, Y.; Okamoto, N.; Toda, K.; Sakamoto, S.; Oben, J.A.; et al. Patatin-like phospholipase domain-containing protein 3 is involved in hepatic fatty acid and triglyceride metabolism through X-box binding protein 1 and modulation of endoplasmic reticulum stress in mice. Hepatol. Res. 2016, 46, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Thoen, L.F.R.; Guimarães, E.L.M.; Dollé, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Borkham-Kamphorst, E.; Stoll, D.; Gressner, A.M.; Weiskirchen, R. Antisense strategy against PDGF B-chain proves effective in preventing experimental liver fibrogenesis. Biochem. Biophys. Res. Commun. 2004, 321, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gea, V.; Ghiassinejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.-P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 552–553. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-W. Lipid droplets, lipophagy, and beyond. Biochim. Biophys. Acta 2016, 1861, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, F.; Czaja, M.J. Regulation and functions of autophagic lipolysis. Trends Endocrinol. Metab. 2016, 27, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Blomhoff, R.; Rasmussen, M.; Nilsson, A.; Norum, K.R.; Berg, T.; Blaner, W.S.; Kato, M.; Mertz, J.R.; Goodman, D.S.; Eriksson, U. Hepatic retinol metabolism. Distribution of retinoids, enzymes, and binding proteins in isolated rat liver cells. J. Biol. Chem. 1985, 260, 13560–13565. [Google Scholar] [PubMed]
- Hautekeete, M.L.; Dodeman, I.; Azais-Braesco, V.; van den Berg, K.; Seynaeve, C.; Geerts, A. Hepatic stellate cells and liver retinoid content in alcoholic liver disease in humans. Alcohol. Clin. Exp. Res. 1998, 22, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Rehm, J.; Samokhvalov, A.V.; Shield, K.D. Global burden of alcoholic liver diseases. J. Hepatol. 2013, 59, 160–168. [Google Scholar] [CrossRef] [PubMed]
- McClain, C.J.; van Thiel, D.H.; Parker, S.; Badzin, L.K.; Gilbert, H. Alterations in Zinc, Vitamin A, and retinol-binding protein in chronic alcoholics: A possible mechanism for night blindness and hypogonadism. Alcohol. Clin. Exp. Res. 1979, 3, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Lieber, C.S. Hepatic vitamin A depletion after chronic ethanol consumption in baboons and rats. J. Nutr. 1981, 111, 2015–2023. [Google Scholar] [PubMed]
- Leo, M.A.; Sato, M.; Lieber, C.S. Effect of hepatic vitamin A depletion on the liver in humans and rats. Gastroenterology 1981, 84, 562–572. [Google Scholar]
- Leo, M.A.; Lieber, C.S. Hepatic vitamin A depletion in alcoholic liver injury. N. Engl. J. Med. 1982, 307, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Bell, H.; Nilsson, A.; Norum, K.R.; Pedersen, L.B.; Raknerud, N.; Rasmussen, M. Retinol and retinyl esters in patients with alcoholic liver disease. J. Hepatol. 1989, 8, 26–31. [Google Scholar] [CrossRef]
- Adachi, S.; Moriwaki, H.; Muto, Y.; Yamada, Y.; Fukutomi, Y.; Shimazaki, M.; Okuno, M.; Ninomiya, M. Reduced retinoid content in hepatocellular carcinoma with special reference to alcohol consumption. Hepatology 1991, 14, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.; Mobarhan, S.; Hupert, J.; Lucchesi, D.; Henderson, C.; Langenberg, P.; Layden, T.J. In vitro stimulation of rat liver retinyl ester hydrolase by ethanol. Arch. Biochem. Biophys. 1989, 269, 69–74. [Google Scholar] [CrossRef]
- Sato, M.; Lieber, C.S. Changes in vitamin A status after acute ethanol administration in the rat. J. Nutr. 1982, 112, 1188–1196. [Google Scholar] [PubMed]
- Vizzutti, F.; Arena, U.; Nobili, V.; Tarquini, R.; Trappoliere, M.; Laffi, G.; Marra, F.; Pinzani, M. Non-invasive assessment of fibrosis in non-alcoholic fatty liver disease (NAFLD). Ann. Hepatol. 2009, 8, 89–94. [Google Scholar] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Younossi, Z.M. Nonalcoholic fatty liver disease: A manifestation of the metabolic syndrome. Clevel. Clin. J. Med. 2008, 75, 721–728. [Google Scholar] [CrossRef]
- Sundaram, S.S.; Zeitler, P.; Nadeau, K. The metabolic syndrome and nonalcoholic fatty liver disease in children. Curr. Opin. Pediatr. 2009, 21, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Chaves, G.V.; Pereira, S.E.; Saboya, C.J.; Spitz, D.; Rodrigues, C.S.; Ramalho, A. Association between liver vitamin A reserves and severity of nonalcoholic fatty liver disease in the class III obese following bariatric surgery. Obes. Surg. 2014, 24, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Palace, V.P.; Khaper, N.; Qin, Q.; Singal, P.K. Antioxidant potentials of vitamin A and carotenoids and their relevance to heart disease. Free Radic. Biol. Med. 1999, 26, 746–761. [Google Scholar] [CrossRef]
- Polimeni, L.; Del Ben, M.; Baratta, F.; Perri, L.; Albanese, F.; Pastori, D.; Violi, F.; Angelico, F. Oxidative stress: New insights on the association of non-alcoholic fatty liver disease and atherosclerosis. World J. Hepatol. 2015, 7, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Reeves, H.L.; Friedman, S.L. Activation of hepatic stellate cells—A key issue in liver fibrosis. Front. Biosci. 2002, 7, d808–d826. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Cytokines and fibrogenesis. Semin. Liver Dis. 1999, 19, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Kovarova, M.; Königsrainer, I.; Königsrainer, A.; Machicao, F.; Häring, H.-U.; Schleicher, E.; Peter, A. The genetic variant I148M in PNPLA3 is associated with increased hepatic retinyl-palmitate storage in humans. J. Clin. Endocrinol. Metab. 2015, 100, E1568–E1574. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, P.; Dongiovanni, P.; Motta, B.M.; Meroni, M.; Lepore, S.M.; Mancina, R.M.; Pelusi, S.; Russo, C.; Caddeo, A.; Rossi, G.; et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum. Mol. Genet. 2016. [Google Scholar] [CrossRef] [PubMed]
- Mondul, A.; Mancina, R.M.; Merlo, A.; Dongiovanni, P.; Rametta, R.; Montalcini, T.; Valenti, L.; Albanes, D.; Romeo, S. PNPLA3 I148M variant influences circulating retinol in adults with nonalcoholic fatty liver disease or obesity. J. Nutr. 2015, 145, 1687–1691. [Google Scholar] [CrossRef] [PubMed]
- Kataria, Y.; Deaton, R.J.; Enk, E.; Jin, M.; Petrauskaite, M.; Dong, L.; Goldenberg, J.R.; Cotler, S.J.; Jensen, D.M.; van Breemen, R.B.; et al. Retinoid and carotenoid status in serum and liver among patients at high-risk for liver cancer. BMC Gastroenterol. 2016, 16, 30. [Google Scholar] [CrossRef] [PubMed]
- Santana, R.C.; Machado, A.A.; Martinelli, A.L.C.; Jordão, A.A.; Ramalho, L.N.Z.; Vannucchi, H. Assessment of indicators of vitamin A status in non-cirrhotic chronic hepatitis C patients. Braz. J. Med. Biol. Res. 2016, 49, e4785. [Google Scholar] [CrossRef] [PubMed]
- Arantes Ferreira Peres, W.; Villaça Chaves, G.; Saraiva Gonçalves, J.C.; Ramalho, A.; Moraes Coelho, H.S. Assessment of the relative dose-response test as indicators of hepatic vitamin A stores in various stages of chronic liver disease. Nutr. Clin. Pract. 2013, 28, 95–100. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grumet, L.; Taschler, U.; Lass, A. Hepatic Retinyl Ester Hydrolases and the Mobilization of Retinyl Ester Stores. Nutrients 2017, 9, 13. https://doi.org/10.3390/nu9010013
Grumet L, Taschler U, Lass A. Hepatic Retinyl Ester Hydrolases and the Mobilization of Retinyl Ester Stores. Nutrients. 2017; 9(1):13. https://doi.org/10.3390/nu9010013
Chicago/Turabian StyleGrumet, Lukas, Ulrike Taschler, and Achim Lass. 2017. "Hepatic Retinyl Ester Hydrolases and the Mobilization of Retinyl Ester Stores" Nutrients 9, no. 1: 13. https://doi.org/10.3390/nu9010013
APA StyleGrumet, L., Taschler, U., & Lass, A. (2017). Hepatic Retinyl Ester Hydrolases and the Mobilization of Retinyl Ester Stores. Nutrients, 9(1), 13. https://doi.org/10.3390/nu9010013