The Role of Carbohydrate Response Element Binding Protein in Intestinal and Hepatic Fructose Metabolism


{kind=link}
{kind=link}
{kind=link}
{kind=link}
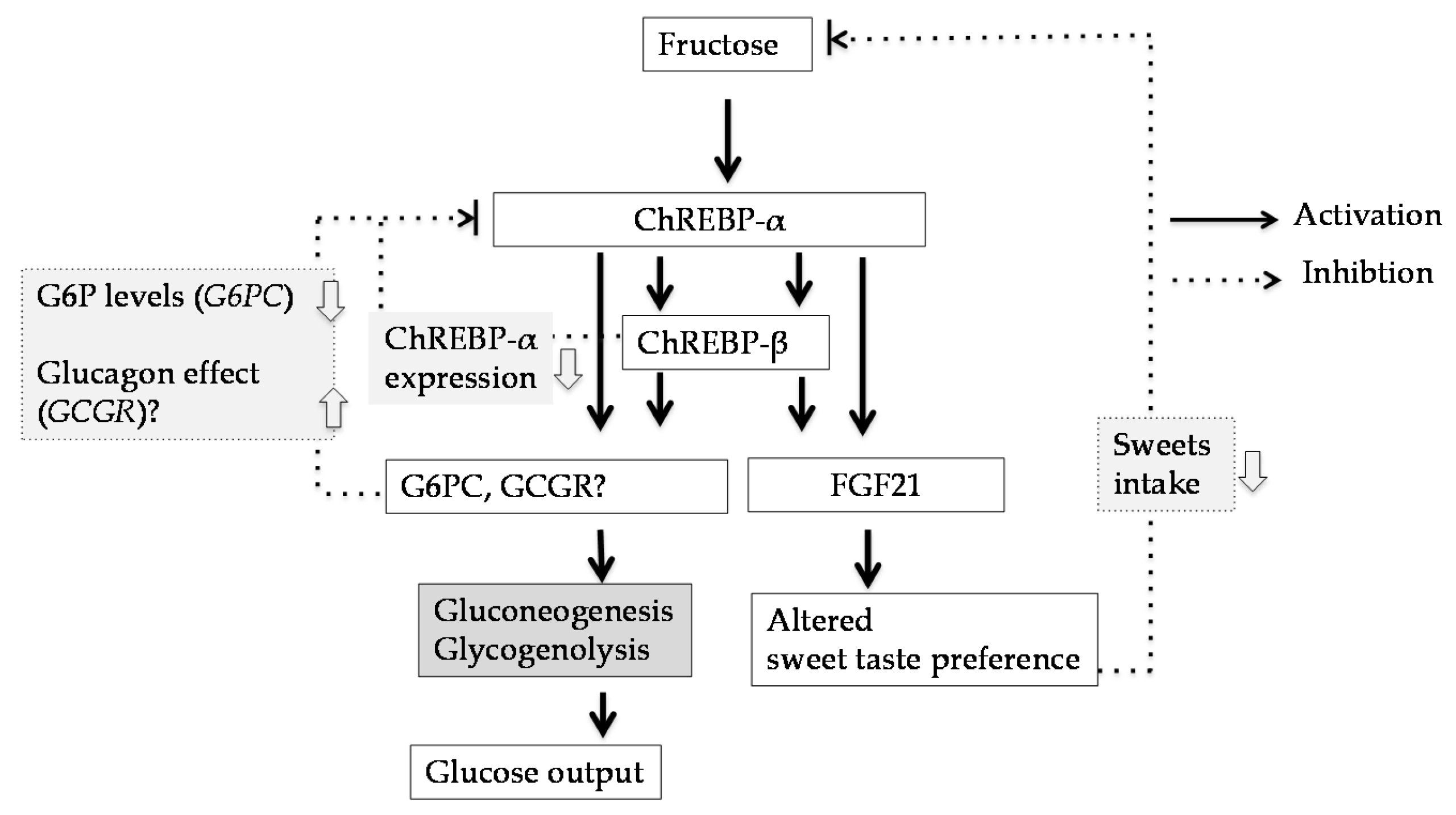{kind=link}
Abstract
:1. Introduction
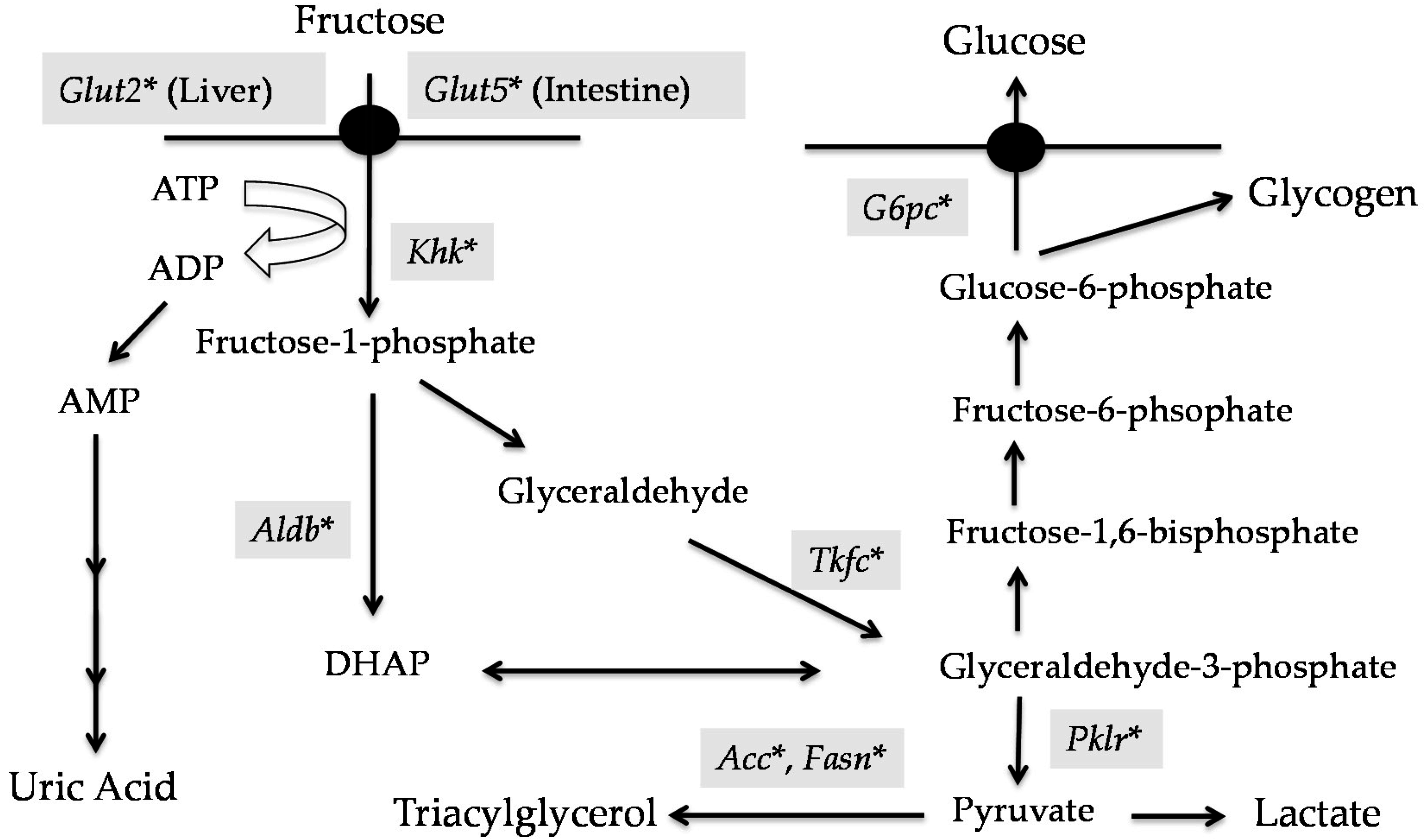
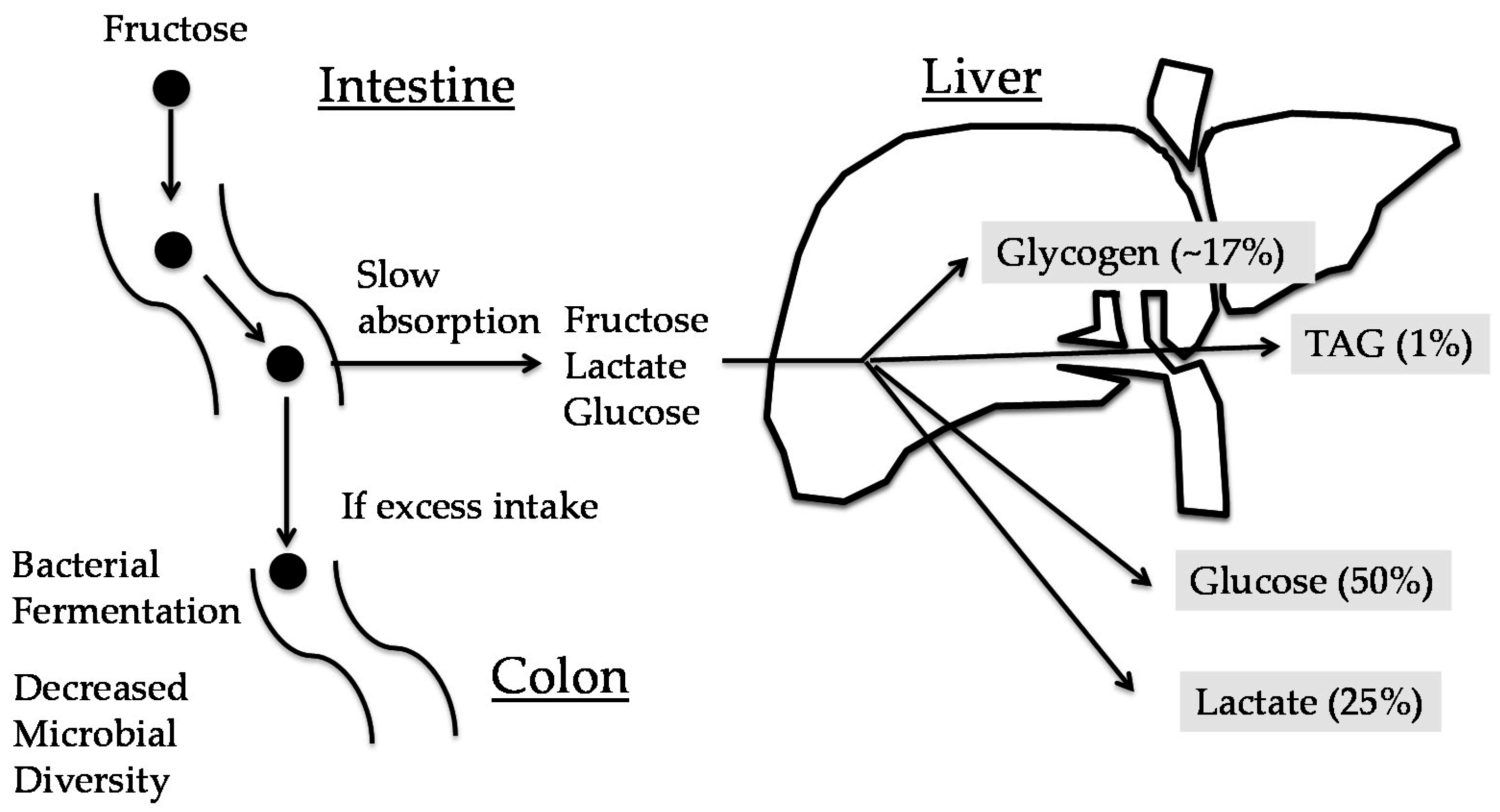
2. Metabolic Fate of Fructose
2.1. The Role of the Intestine in Fructose Metabolism
2.2. Role of the Liver in Fructose Metabolism
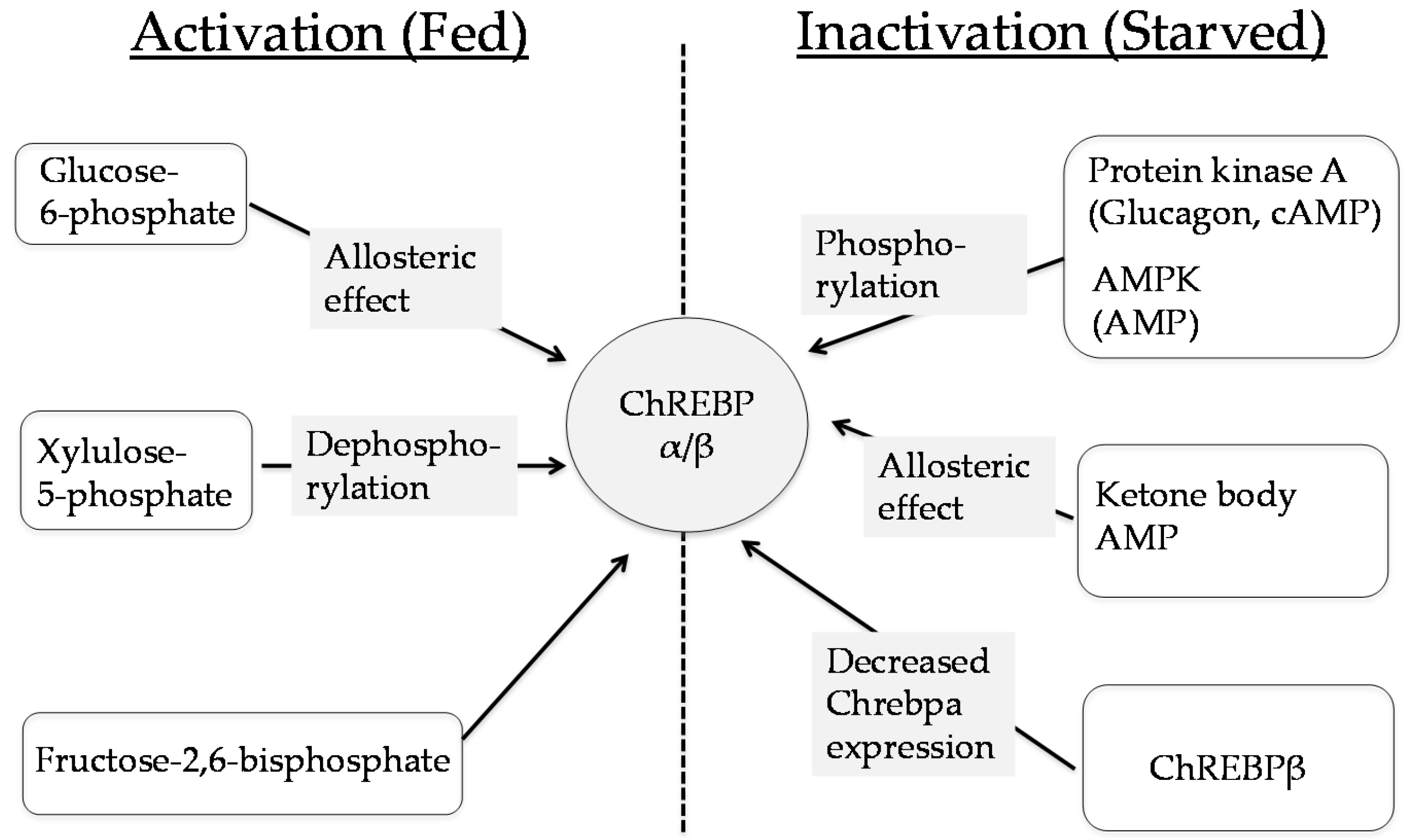
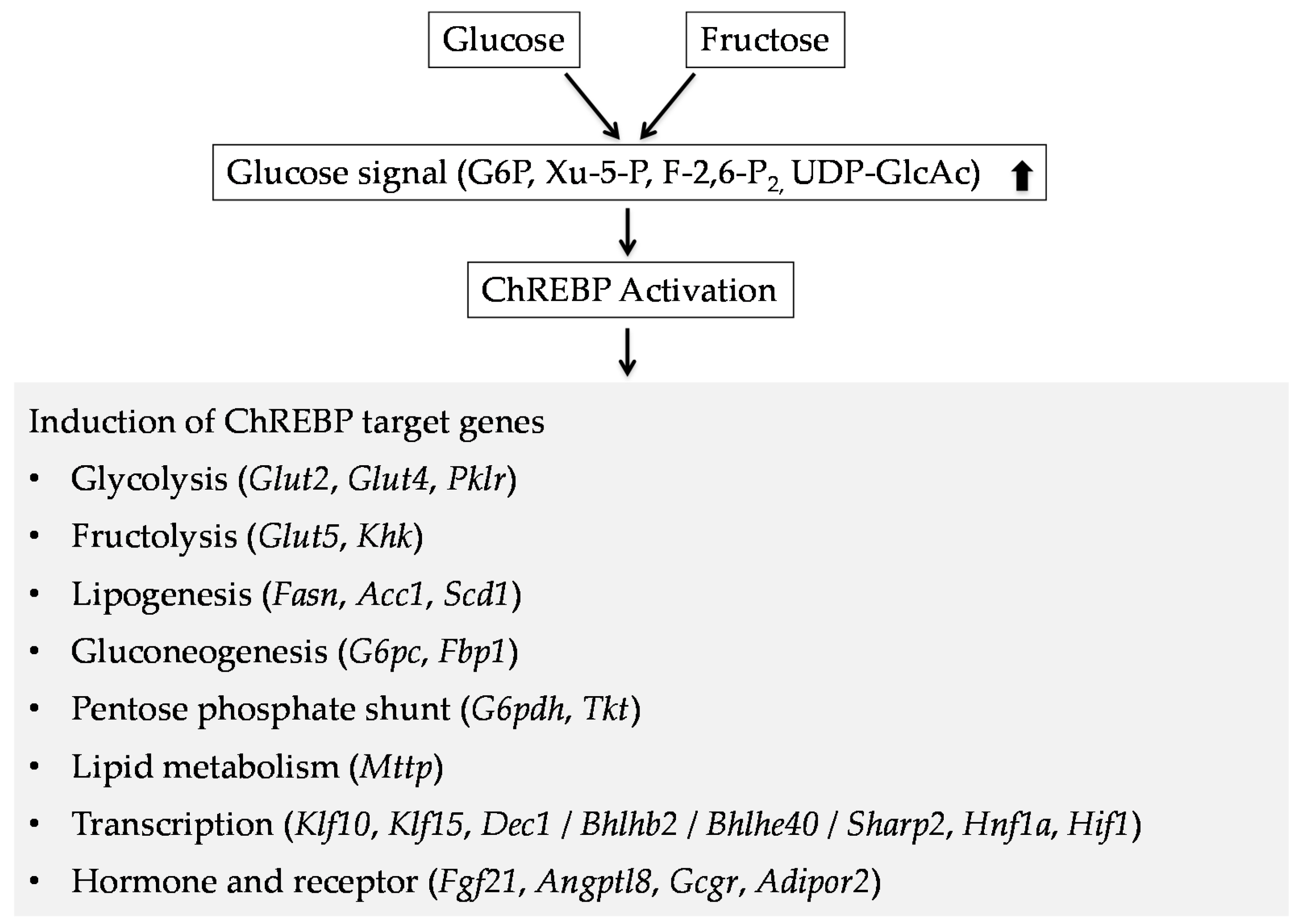
3. ChREBP Regulates Glycolysis and Fructolysis through Altered Gene Expression
3.1. Glucose Metabolism
3.2. Fructose Metabolism
4. Chrebp Deletion Suppresses Obesity and Fatty Liver Induced by Excess Carbohydrate Feeding
5. Newly-Identified Roles of ChREBP: Regulation of Sweet Preference and Hepatic Glucose Production
5.1. Fibroblast Growth Factor (FGF)-21
5.2. Glucose-6-Phosphatase Catalytic Subunit (G6pc)
6. The Role of ChREBP in Fructose Metabolism
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Macdonald, I.A. A review of recent evidence relating to sugars, insulin resistance and diabetes. Eur. J. Nutr. 2016, 55, 17–23. [Google Scholar] [CrossRef]
- Khan, T.A.; Sievenpiper, J.L. Controversies about sugars: Results from systematic reviews and meta-analyses on obesity, cardiometabolic disease and diabetes. Eur. J. Nutr. 2016, 55, 25–43. [Google Scholar] [PubMed]
- Elliott, S.S.; Keim, N.L.; Stern, J.S.; Teff, K.; Havel, P.J. Fructose, weight gain, and the insulin resistance syndrome. Am. J. Clin. Nutr. 2002, 76, 911–922. [Google Scholar] [PubMed]
- Samuel, V.T. Fructose induced lipogenesis: From sugar to fat to insulin resistance. Trends Endocrinol. Metab. 2011, 22, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Samuel, V.T. The Sweet Path to Metabolic Demise: Fructose and Lipid Synthesis. Trends Endocrinol. Metab. 2016, 27, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Hosotani, T.; Kawasaki, T.; Nakagawa, K.; Hayashi, S.; Nakano, Y.; Inui, H.; Yamanouchi, T. Eucalyptus leaf extract suppresses the postprandial elevation of portal, cardiac and peripheral fructose concentrations after sucrose ingestion in rats. J. Clin. Biochem. Nutr. 2010, 46, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Akanuma, H.; Yamanouchi, T. Increased fructose concentrations in blood and urine in patients with diabetes. Diabetes Care 2002, 25, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, C.G.; Stehouwer, C.D.; van Hinsbergh, V.W. Fructose-mediated non-enzymatic glycation: Sweet coupling or bad modification. Diabetes Metab. Res. Rev. 2004, 20, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, L.M.; Benson, V.L.; Ritchie, R.H.; Mellor, K.M. Diabetic Cardiomyopathy: The Case for a Role of Fructose in Disease Etiology. Diabetes 2016, 65, 3521–3528. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Lê, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Z.; Empie, M.W. Fructose metabolism in humans—What isotopic tracer studies tell us. Nutr. Metab. 2012, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Douard, V.; Ferraris, R.P. The role of fructose transporters in diseases linked to excessive fructose intake. J Physiol. 2013, 591, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Ebert, K.; Witt, H. Fructose malabsorption. Mol. Cell. Pediatr. 2016, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K. The transcription factor carbohydrate-response element-binding protein (ChREBP): A possible link between metabolic disease and cancer. Biochim. Biophys. Acta 2016, 1863, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Uyeda, K.; Repa, J.J. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006, 4, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Filhoulaud, G.; Guilmeau, S.; Dentin, R.; Girard, J.; Postic, C. Novel insights into ChREBP regulation and function. Trends Endocrinol. Metab. 2013, 24, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Towle, H.C. Glucose as a regulator of eukaryotic gene transcription. Trends Endocrinol. Metab. 2005, 16, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Takenoshita, M.; Sakurai, M.; Bruick, R.K.; Henzel, W.J.; Shillinglaw, W.; Arnot, D.; Uyeda, K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. USA 2001, 98, 9116–9121. [Google Scholar] [CrossRef] [PubMed]
- Cairo, S.; Merla, G.; Urbinati, F.; Ballabio, A.; Reymond, A. WBSCR14, a gene mapping to the Williams--Beuren syndrome deleted region, is a new member of the Mlx transcription factor network. Hum. Mol. Genet. 2001, 10, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Krawczyk, S.A.; Doridot, L.; Fowler, A.J.; Wang, J.X.; Trauger, S.A.; Noh, H.L.; Kang, H.J.; Meissen, J.K.; Blatnik, M.; et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J. Clin. Investig. 2016, 126, 4372–4386. [Google Scholar] [CrossRef] [PubMed]
- Fisher, F.M.; Kim, M.; Doridot, L.; Cunniff, J.C.; Parker, T.S.; Levine, D.M.; Hellerstein, M.K.; Hudgins, L.C.; Maratos-Flier, E.; Herman, M.A. A critical role for ChREBP-mediated FGF21 secretion in hepatic fructose metabolism. Mol. Metab. 2017, 6, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Robinson, L.N.; Towle, H.C. ChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liver. J. Biol. Chem. 2006, 281, 28721–28730. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.S.; Kim, D.; Lee, Y.S.; Kim, H.J.; Han, J.Y.; Im, S.S.; Chong, H.K.; Kwon, J.K.; Cho, Y.H.; Kim, W.K.; et al. Integrated expression profiling and genome-wide analysis of ChREBP targets reveals the dual role for ChREBP in glucose-regulated gene expression. PLoS ONE 2011, 6, e22544. [Google Scholar] [CrossRef] [PubMed]
- Poungvarin, N.; Chang, B.; Imamura, M.; Chen, J.; Moolsuwan, K.; Sae-Lee, C.; Li, W.; Chan, L. Genome-Wide Analysis of ChREBP Binding Sites on Male Mouse Liver and White Adipose Chromatin. Endocrinology 2015, 156, 1982–1994. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.; Douard, V.; Yu, S.; Gao, N.; Ferraris, R.P. Transport, metabolism, and endosomal trafficking-dependent regulation of intestinal fructose absorption. FASEB J. 2015, 29, 4046–4058. [Google Scholar] [CrossRef] [PubMed]
- Douard, V.; Ferraris, R.P. Regulation of the fructose transporter GLUT5 in health and disease. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E227–E237. [Google Scholar] [CrossRef] [PubMed]
- Latulippe, M.E.; Skoog, S.M. Fructose Malabsorption and Intolerance: Effects of Fructose with and without Simultaneous Glucose Ingestion. Crit. Rev. Food Sci. Nutr. 2011, 51, 583–592. [Google Scholar] [CrossRef] [PubMed]
- DiNicolantonio, J.J.; Lucan, S.C. Is Fructose Malabsorption a Cause of Irritable Bowel Syndrome? Med. Hypotheses 2015, 85, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Riby, J.; Kretchmer, N. Intestinal absorption of fructose in the rat. Gastroenterology 1991, 101, 360–367. [Google Scholar] [CrossRef]
- Barone, S.; Fussell, S.L.; Singh, A.K.; Lucas, F.; Xu, J.; Kim, C.; Wu, X.; Yu, Y.; Amlal, H.; Seidler, U.; et al. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J. Biol. Chem. 2009, 284, 5056–5066. [Google Scholar] [CrossRef] [PubMed]
- Diggle, C.P.; Shires, M.; Leitch, D.; Brooke, D.; Carr, I.M.; Markham, A.F.; Hayward, B.E.; Asipu, A.; Bonthron, D.T. Ketohexokinase: Expression and Localization of the Principal Fructose-metabolizing Enzyme. J. Histochem. Cytochem. 2009, 57, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Hayward, B.E.; Bonthron, D.T. Structure and alternative splicing of the ketohexokinase gene. Eur. J. Biochem. 1998, 257, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Lanaspa, M.A.; Le, M.T.; Garcia, G.E.; Diggle, C.P.; Maclean, P.S.; Jackman, M.R.; Asipu, A.; Roncal-Jimenez, C.A.; Kosugi, T.; et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 4320–4325. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qian, X.; Peng, L.X.; Jiang, Y.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Lee, J.H.; Cote, G.; Wang, H.; et al. A splicing switch from ketohexokinase-C to ketohexokinase-A drives hepatocellular carcinoma formation. Nat. Cell Biol. 2016, 18, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Iizuka, K.; Miller, B.C.; Uyeda, K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 15597–15602. [Google Scholar] [CrossRef] [PubMed]
- Stoeckman, A.K.; Ma, L.; Towle, H.C. Mlx is the functional heteromeric partner of the carbohydrate response element-binding protein in glucose regulation of lipogenic enzyme genes. J. Biol. Chem. 2004, 279, 15662–15669. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schön, M.R.; Abumrad, N.A.; Blüher, M.; Klein, S.; Kahn, B.B. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012, 484, 333–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.V.; Chang, B.; Imamura, M.; Poungvarin, N.; Chan, L. Glucose-dependent transcriptional regulation by an evolutionarily conserved glucose-sensing module. Diabetes 2006, 55, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Jing, G.; Chen, J.; Xu, G.; Shalev, A. Islet ChREBP-β is increased in diabetes and controls ChREBP-α and glucose-induced gene expression via a negative feedback loop. Mol. Metab. 2016, 5, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Wu, W.; Horikawa, Y.; Saito, M.; Takeda, J. Feedback looping between ChREBP and PPARα in the regulation of lipid metabolism in brown adipose tissues. Endocr. J. 2013, 60, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Wu, W.; Horikawa, Y.; Takeda, J. Role of glucose-6-phosphate and xylulose-5-phosphate in the regulation of glucose-stimulated gene expression in the pancreatic β cell line, INS-1E. Endocr. J. 2013, 60, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Kabashima, T.; Kawaguchi, T.; Wadzinski, B.E.; Uyeda, K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc. Natl. Acad. Sci. USA 2003, 100, 5107–5112. [Google Scholar] [CrossRef] [PubMed]
- Li, M.V.; Chen, W.; Harmancey, R.N.; Nuotio-Antar, A.M.; Imamura, M.; Saha, P.; Taegtmeyer, H.; Chan, L. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP). Biochem. Biophys. Res. Commun. 2010, 395, 395–400. [Google Scholar] [CrossRef]
- Dentin, R.; Tomas-Cobos, L.; Foufelle, F.; Leopold, J.; Girard, J.; Postic, C.; Ferré, P. Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in the liver. J. Hepatol. 2012, 56, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Arden, C.; Tudhope, S.J.; Petrie, J.L.; Al-Oanzi, Z.H.; Cullen, K.S.; Lange, A.J.; Towle, H.C.; Agius, L. Fructose 2,6-bisphosphate is essential for glucose-regulated gene transcription of glucose-6-phosphatase and other ChREBP target genes in hepatocytes. Biochem. J. 2012, 443, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Ido-Kitamura, Y.; Sasaki, T.; Kobayashi, M.; Kim, H.J.; Lee, Y.S.; Kikuchi, O.; Yokota-Hashimoto, H.; Iizuka, K.; Accili, D.; Kitamura, T. Hepatic FoxO1 integrates glucose utilization and lipid synthesis through regulation of Chrebp O-glycosylation. PLoS ONE 2012, 7, e47231. [Google Scholar] [CrossRef] [PubMed]
- Guinez, C.; Filhoulaud, G.; Rayah-Benhamed, F.; Marmier, S.; Dubuquoy, C.; Dentin, R.; Moldes, M.; Burnol, A.F.; Yang, X.; Lefebvre, T.; et al. GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 2011, 60, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Osatomi, K.; Yamashita, H.; Kabashima, T.; Uyeda, K. Mechanism for fatty acid “sparing” effect on glucose-induced transcription: Regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J. Biol. Chem. 2002, 277, 3829–3835. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Ge, Q.; Pawlosky, R.; Wynn, R.M.; Veech, R.L.; Uyeda, K. Metabolite regulation of nucleo-cytosolic trafficking of carbohydrate response element-binding protein (ChREBP): Role of ketone bodies. J. Biol. Chem. 2013, 288, 28358–28367. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Jung, H.; Nakagawa, T.; Pawlosky, R.; Takeshima, T.; Lee, W.R.; Sakiyama, H.; Laxman, S.; Wynn, R.M.; Tu, B.P.; et al. Metabolite regulation of nuclear localization of carbohydrate-response element-binding protein (ChREBP): Role of AMP as an allosteric inhibitor. J. Biol. Chem. 2016, 291, 10515–10527. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Takenoshita, M.; Kabashima, T.; Uyeda, K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. USA 2001, 98, 13710–13715. [Google Scholar] [CrossRef] [PubMed]
- Matosin-Matekalo, M.; Mesonero, J.E.; Laroche, T.J.; Lacasa, M.; Brot-Laroche, E. Glucose and thyroid hormone co-regulate the expression of the intestinal fructose transporter GLUT5. Biochem. J. 1999, 339, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, K.; Sakaguchi, N.; Goda, T. Triiodothyronine (T3) and fructose coordinately enhance expression of the GLUT5 gene in the small intestine of rats during weaning period. Biosci. Biotechnol. Biochem. 2007, 71, 1345–1347. [Google Scholar] [CrossRef] [PubMed]
- Gouyon, F.; Onesto, C.; Dalet, V.; Pages, G.; Leturque, A.; Brot-Laroche, E. Fructose modulates GLUT5 mRNA stability in differentiated Caco-2 cells: Role of cAMP-signalling pathway and PABP (polyadenylated-binding protein)-interacting protein (Paip) 2. Biochem. J. 2003, 375, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Shalev, A. Keeping tabs on fructose. Elife 2016, 5, e21263. [Google Scholar] [CrossRef] [PubMed]
- Dotimas, J.R.; Lee, A.W.; Schmider, A.B.; Carroll, S.H.; Shah, A.; Bilen, J.; Elliott, K.R.; Myers, R.B.; Soberman, R.J.; Yoshioka, J.; et al. Diabetes regulates fructose absorption through thioredoxin-interacting protein. Elife 2016, 5, e18313. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, E.; Masaki, S.; Matsuo, Y.; Chen, Z.; Tian, H.; Yodoi, J. Thioredoxin/Txnip: Redoxisome, as a redox switch for the pathogenesis of diseases. Front. Immunol. 2014, 4, 514. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.W.; Stoltzman, C.A.; Sighinolfi, M.P.; Han, K.S.; Ayer, D.E. Glucose controls nuclear accumulation, promoter binding, and transcriptional activity of the MondoA-Mlx heterodimer. Mol. Cell Biol. 2010, 30, 2887–2895. [Google Scholar] [CrossRef] [PubMed]
- Cha-Molstad, H.; Saxena, G.; Chen, J.; Shalev, A. Glucose-stimulated expression of Txnip is mediated by carbohydrate response element-binding protein, p300, and histone H4 acetylation in pancreatic beta cells. J. Biol. Chem. 2009, 284, 16898–16905. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Cicerchi, C.; Li, N.; Roncal-Jimenez, C.A.; Ishimoto, T.; Le, M.; Garcia, G.E.; Thomas, J.B.; Rivard, C.J.; et al. Uric Acid Stimulates Fructokinase and Accelerates Fructose Metabolism in the Development of Fatty Liver. PLoS ONE 2012, 7, e47948. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.; Douard, V.; Yu, S.; Tharabenjasin, P.; Gao, N.; Ferraris, R.P. Fructose-induced increases in expression of intestinal fructolytic and gluconeogenic genes are regulated by GLUT5 and KHK. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R499–R509. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Tsuchida, H.; Kato, T.; Niwa, H.; Horikawa, Y.; Takeda, J.; Iizuka, K. Fat and carbohydrate in western diet contribute differently to hepatic lipid accumulation. Biochem. Biophys. Res. Commun. 2015, 461, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Miller, B.; Uyeda, K. Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E358–E364. [Google Scholar] [CrossRef] [PubMed]
- Dentin, R.; Benhamed, F.; Hainault, I.; Fauveau, V.; Foufelle, F.; Dyck, J.R.; Girard, J.; Postic, C. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 2006, 55, 2159–2170. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J. FGF21 and metabolic disease in 2016: A new frontier in FGF21 biology. Nat. Rev. Endocrinol. 2017, 13, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Takeda, J.; Horikawa, Y. Glucose induces FGF21 mRNA expression through ChREBP activation in rat hepatocytes. FEBS Lett. 2009, 583, 2882–2886. [Google Scholar] [CrossRef] [PubMed]
- Dushay, J.R.; Toschi, E.; Mitten, E.K.; Fisher, F.M.; Herman, M.A.; Maratos-Flier, E. Fructose ingestion acutely stimulates circulating FGF21 levels in humans. Mol. Metab. 2014, 4, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Lin, V.Y.; Goetz, R.; Mohammadi, M.; Mangelsdorf, D.J.; Kliewer, S.A. Inhibition of growth hormone signaling by the fasting-induced hormone FGF21. Cell Metab. 2008, 8, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Von Holstein-Rathlou, S.; BonDurant, L.D.; Peltekian, L.; Naber, M.C.; Yin, T.C.; Claflin, K.E.; Urizar, A.I.; Madsen, A.N.; Ratner, C.; Holst, B.; et al. FGF21 Mediates Endocrine Control of Simple Sugar Intake and Sweet Taste Preference by the Liver. Cell Metab. 2016, 23, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Owen, B.M.; Song, P.; Hernandez, G.; Zhang, Y.; Zhou, Y.; Scott, W.T.; Paratala, B.; Turner, T.; Smith, A.; et al. FGF21 Regulates Sweet and Alcohol Preference. Cell Metab. 2016, 23, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.B.; Zhang, P.; Doumen, C.; Charbonnet, M.; Lu, D.; Newgard, C.B.; Haycock, J.W.; Lange, A.J.; Scott, D.K. The promoter for the gene encoding the catalytic subunit of rat glucose-6-phosphatase contains two distinct glucose-responsive regions. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E788–E801. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Tomita, R.; Takeda, J.; Horikawa, Y. Rat glucagon receptor mRNA is directly regulated by glucose through transactivation of the carbohydrate response element binding protein. Biochem. Biophys. Res. Commun. 2012, 417, 1107–1112. [Google Scholar] [CrossRef]
- Eissing, L.; Scherer, T.; Tödter, K.; Knippschild, U.; Greve, J.W.; Buurman, W.A.; Pinnschmidt, H.O.; Rensen, S.S.; Wolf, A.M.; Bartelt, A.; et al. De novo lipogenesis in human fat and liver is linked to ChREBP-β and metabolic health. Nat. Commun. 2013, 4, 1528. [Google Scholar] [CrossRef] [PubMed]
- Halmos, E.P.; Power, V.A.; Shepherd, S.J.; Gibson, P.R.; Muir, J.G. A diet low in FODMAPs reduces symptoms of irritable bowel syndrome. Gastroenterology 2014, 146, 67.e5–75.e5. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iizuka, K. The Role of Carbohydrate Response Element Binding Protein in Intestinal and Hepatic Fructose Metabolism. Nutrients 2017, 9, 181. https://doi.org/10.3390/nu9020181
Iizuka K. The Role of Carbohydrate Response Element Binding Protein in Intestinal and Hepatic Fructose Metabolism. Nutrients. 2017; 9(2):181. https://doi.org/10.3390/nu9020181
Chicago/Turabian StyleIizuka, Katsumi. 2017. "The Role of Carbohydrate Response Element Binding Protein in Intestinal and Hepatic Fructose Metabolism" Nutrients 9, no. 2: 181. https://doi.org/10.3390/nu9020181
APA StyleIizuka, K. (2017). The Role of Carbohydrate Response Element Binding Protein in Intestinal and Hepatic Fructose Metabolism. Nutrients, 9(2), 181. https://doi.org/10.3390/nu9020181