Prenatal Exposure to a Maternal High-Fat Diet Affects Histone Modification of Cardiometabolic Genes in Newborn Rats

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
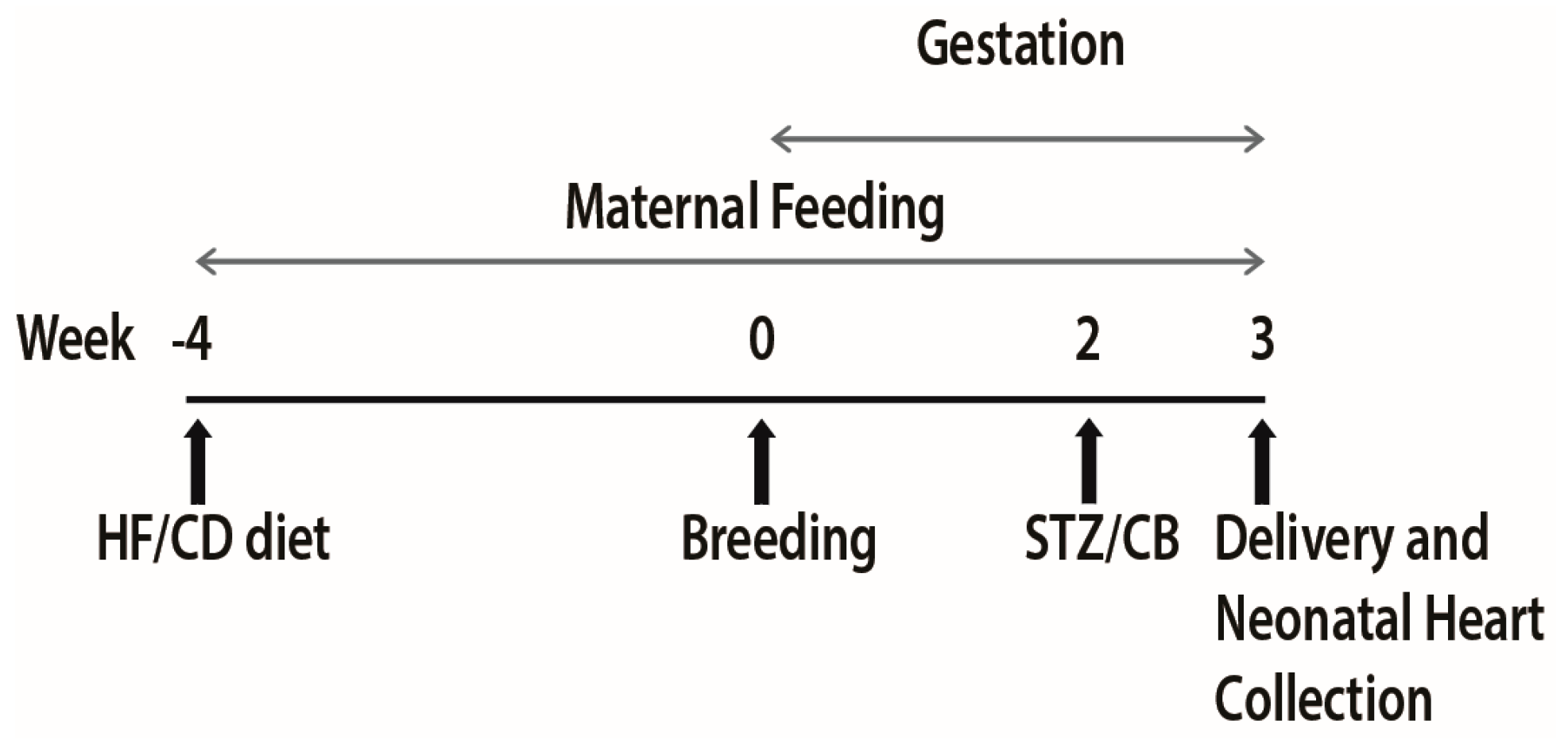
2.1. Animal Study and Dietary Intervention
2.2. Chromatin Immunoprecipitation (ChIP) Assay
2.3. ChIP Sequencing
2.4. Bioinformatics Analyses
2.5. RNA Isolation and RT-PCR
2.6. Statistical Analyses
3. Results
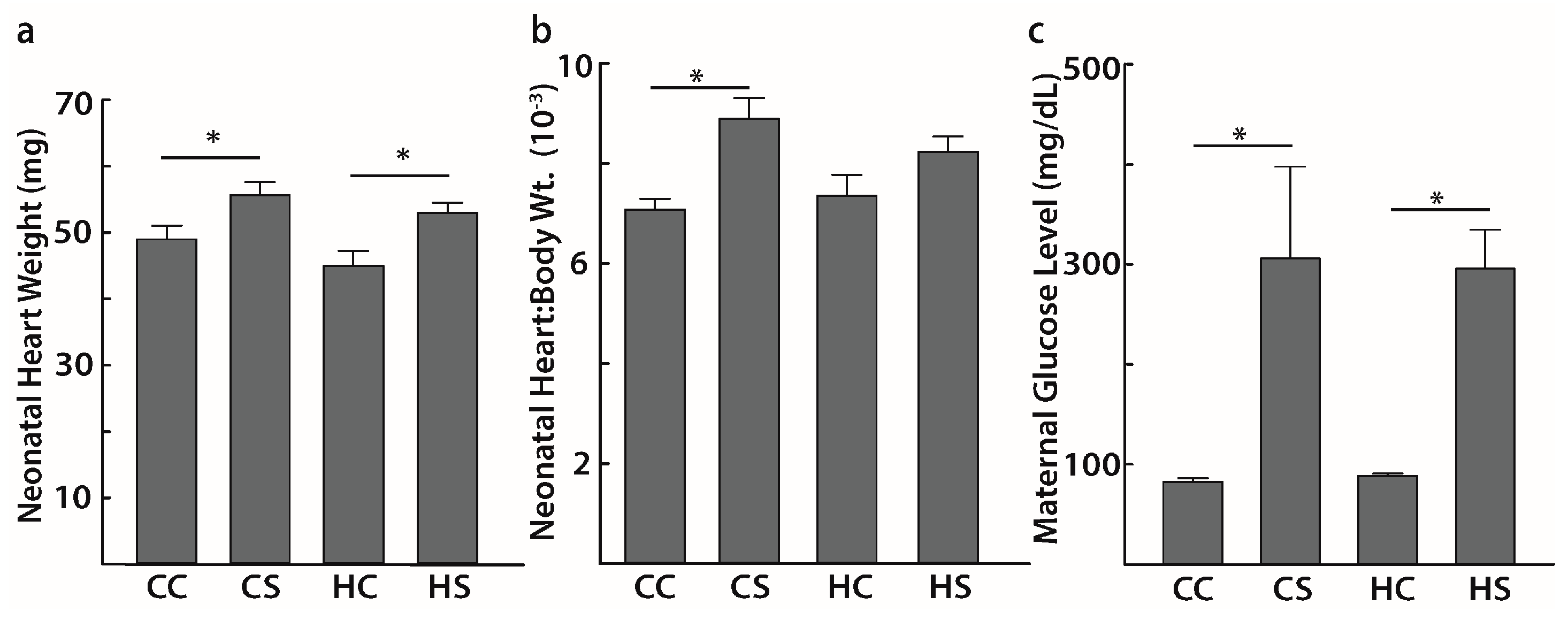
3.1. Maternal Late Gestation Diabetes Increases Heart Weight in Offspring
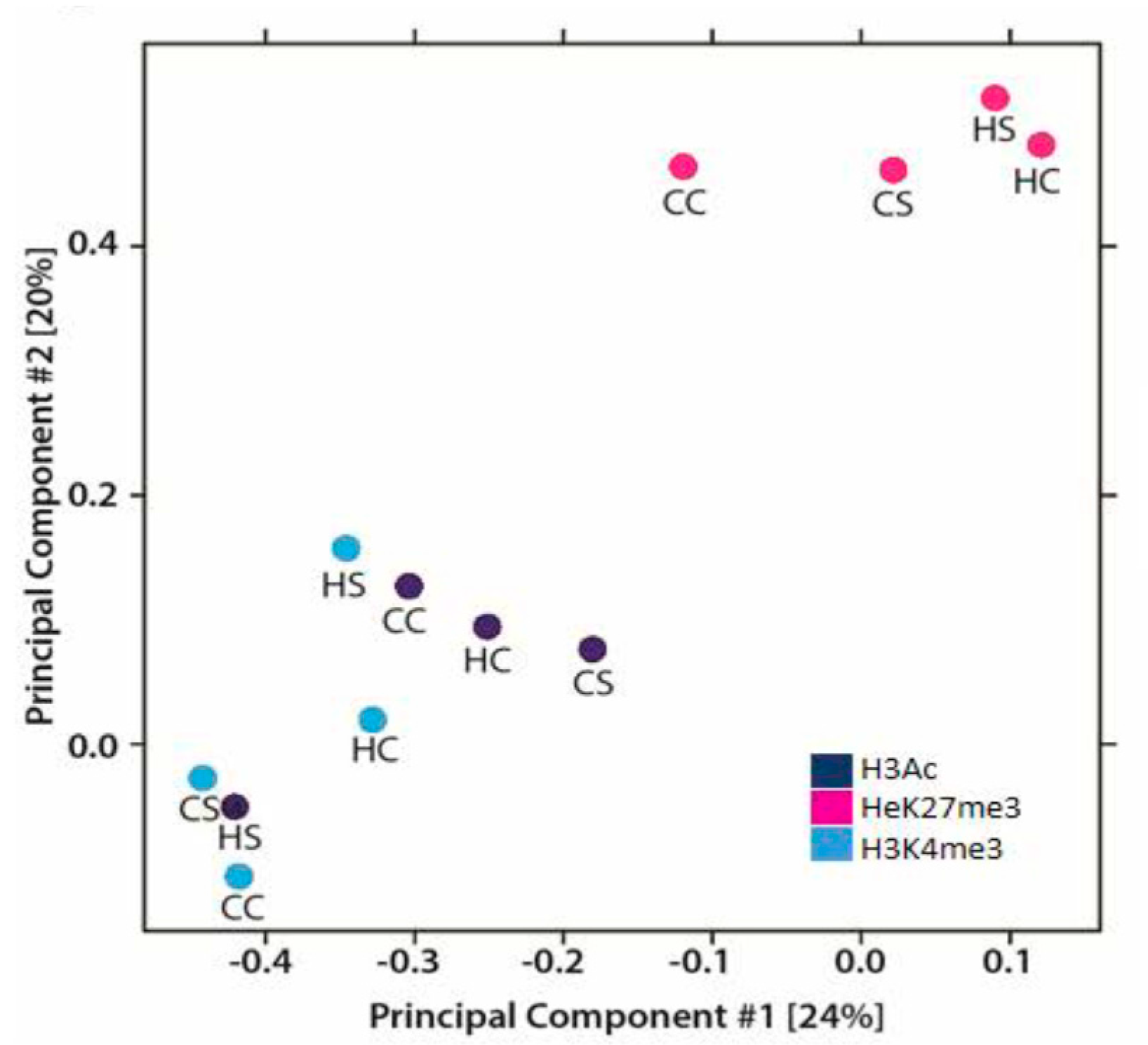
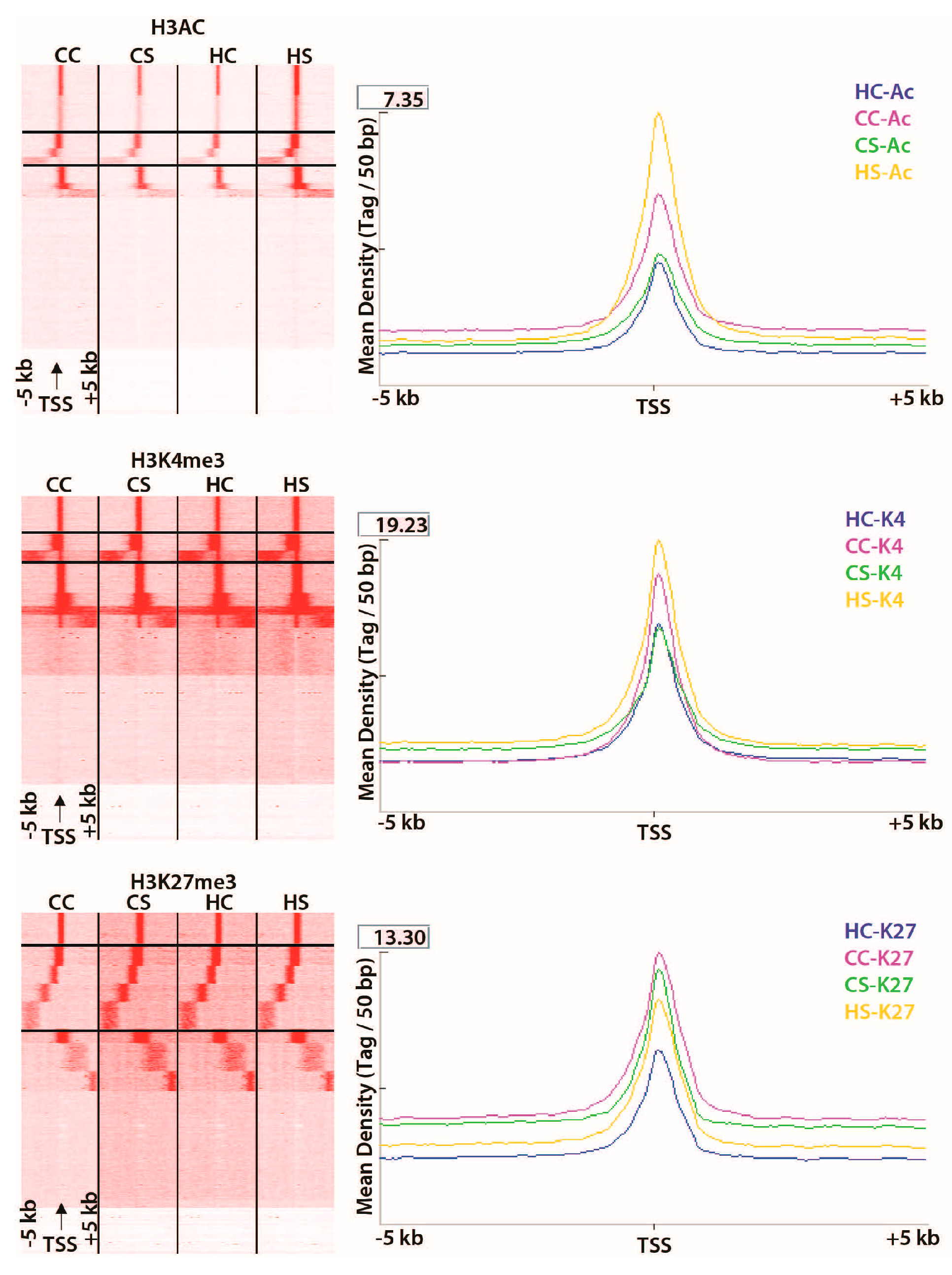
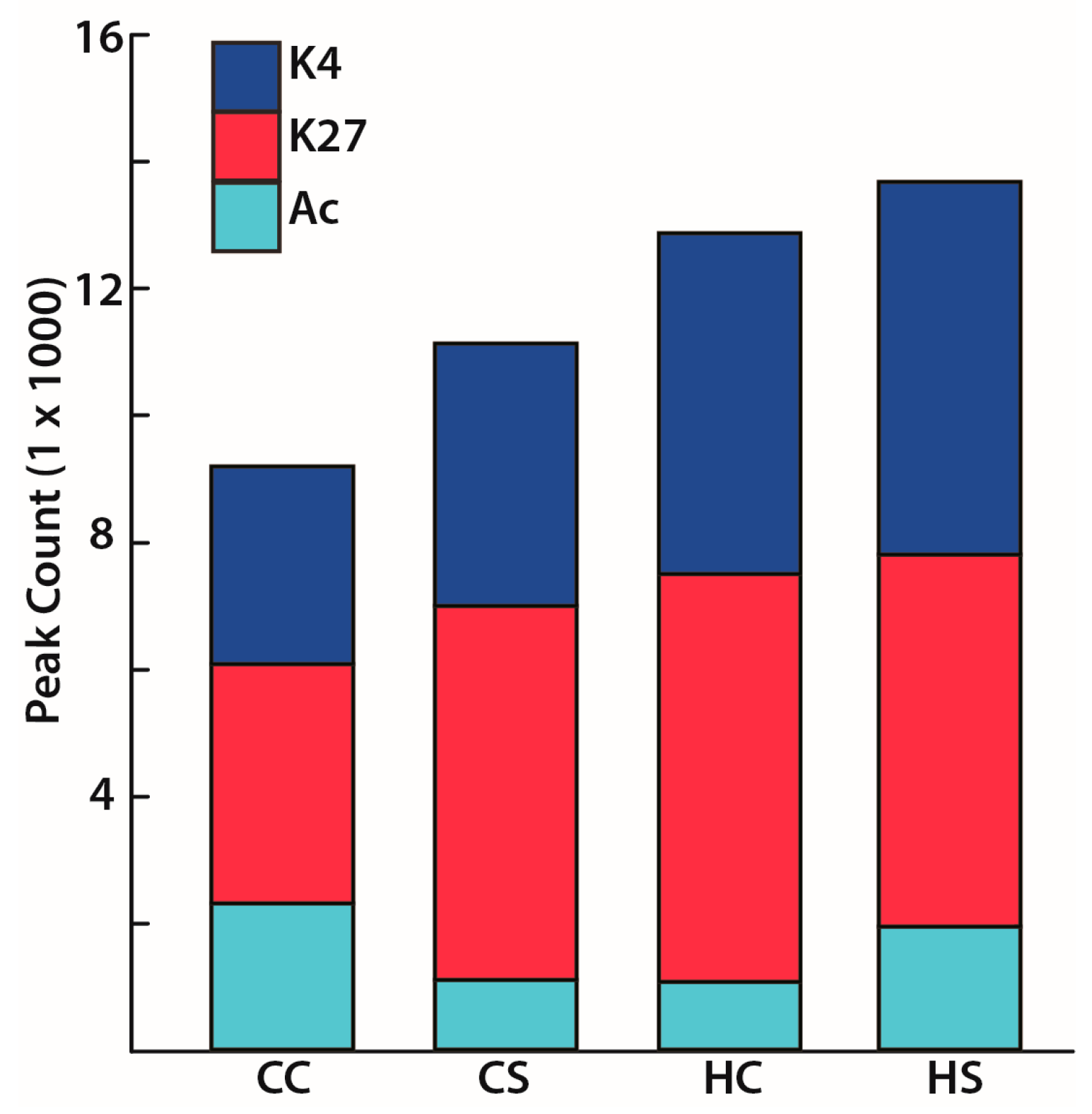
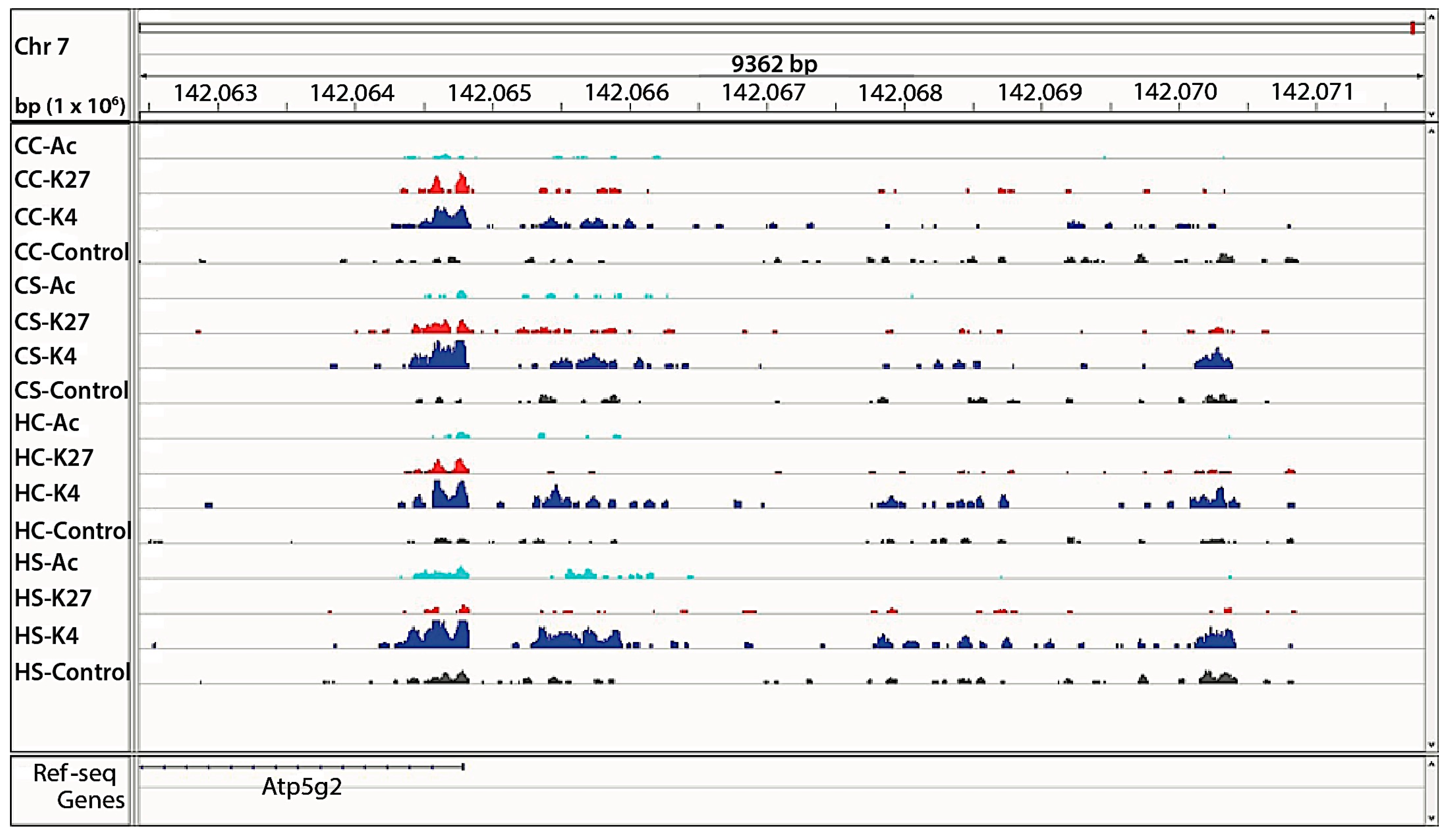
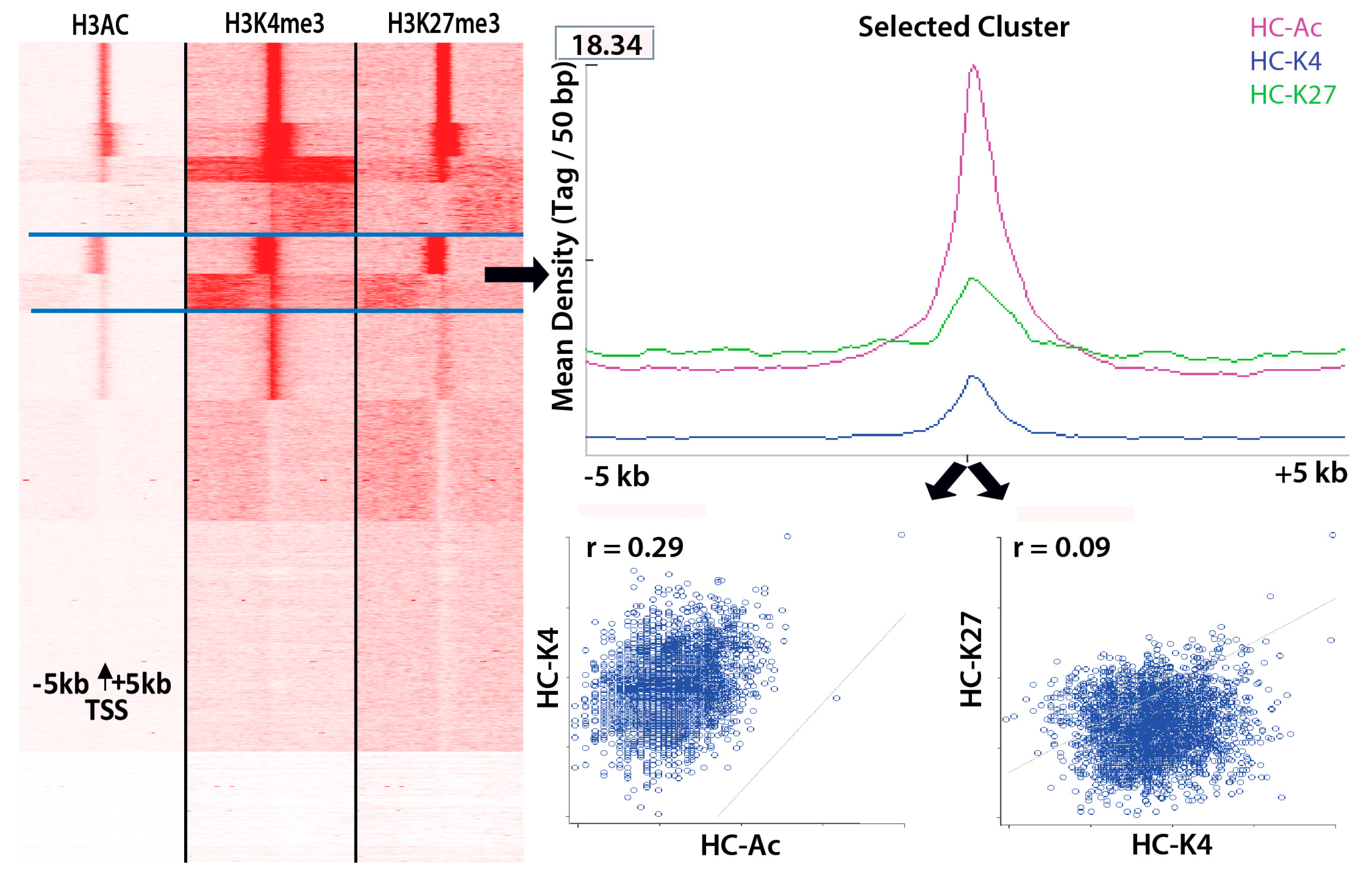
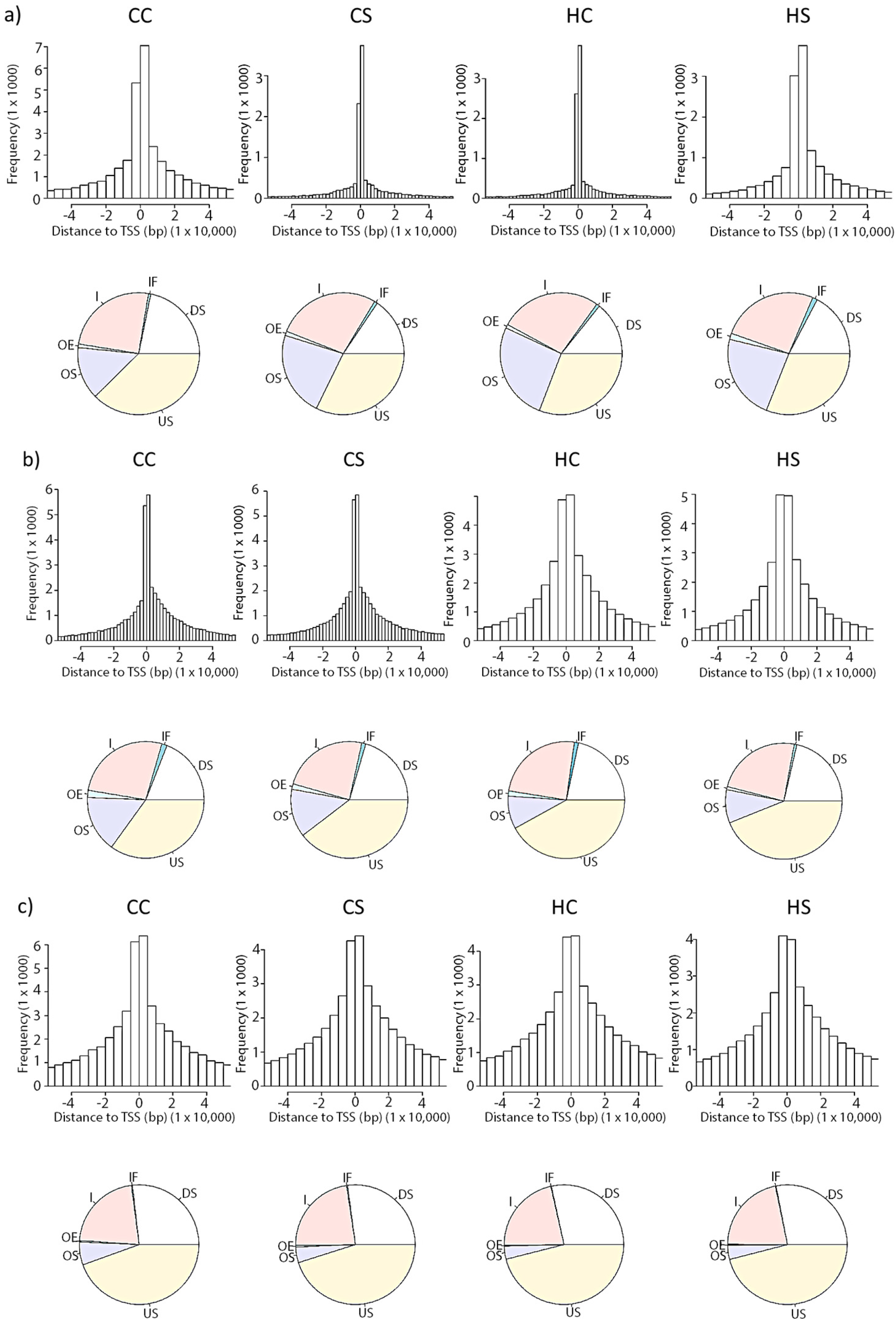
3.2. In Utero Exposure to Diabetes or HF-Diet Associates with Differential Histone Modifications in Offspring Cardiac Tissue
3.3. Differential Binding Analyses of Diabetes and Diet Induced Histone Modifications
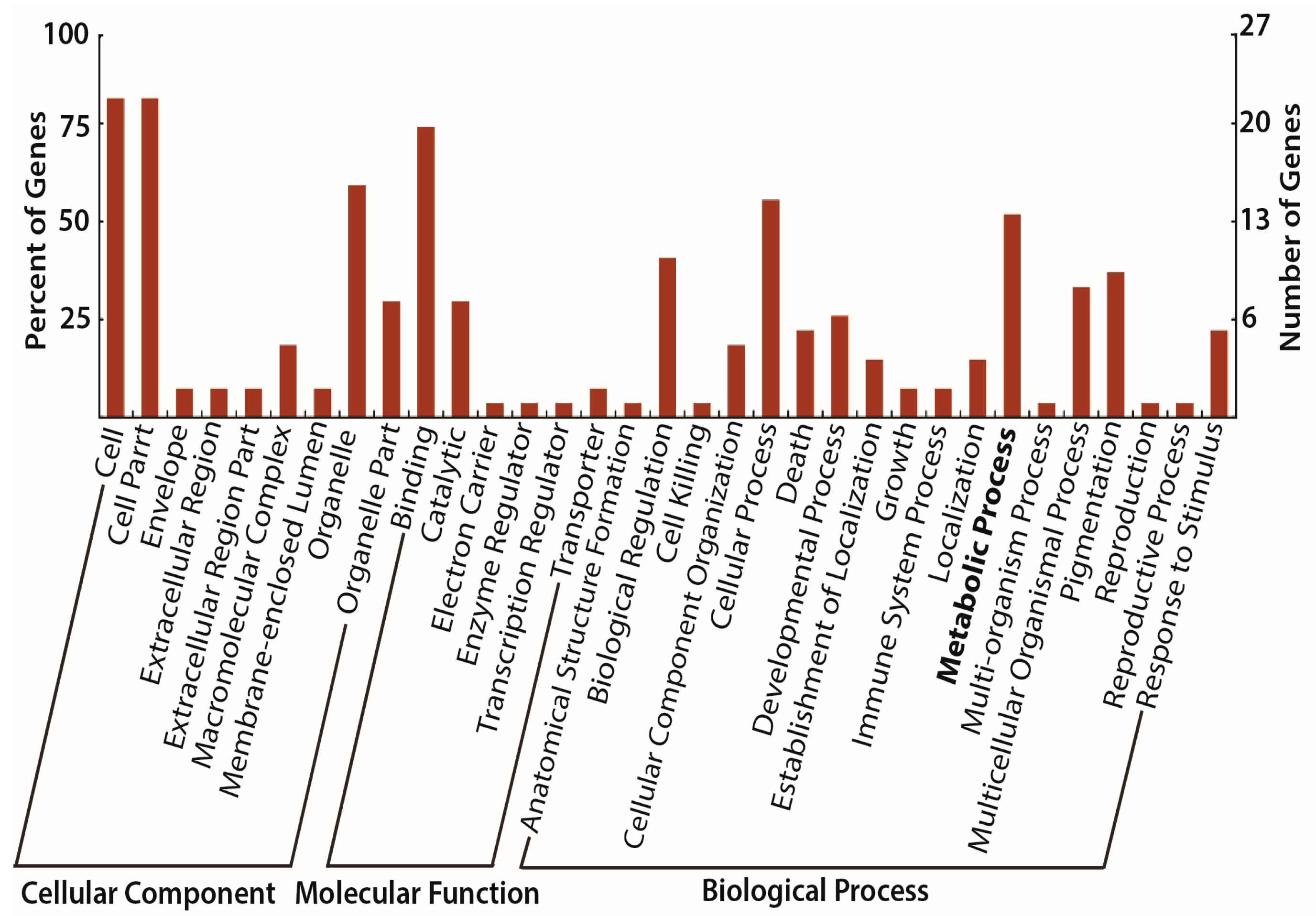
3.4. Gene Promoter Occupancy of Diabetes and Diet Induced Differential Histone Modifications
3.5. Quantitative Association of Diet and Diabetes Induced Histone Modifications with Candidate Genes and QTLs Related to Cardiometabolic Functions
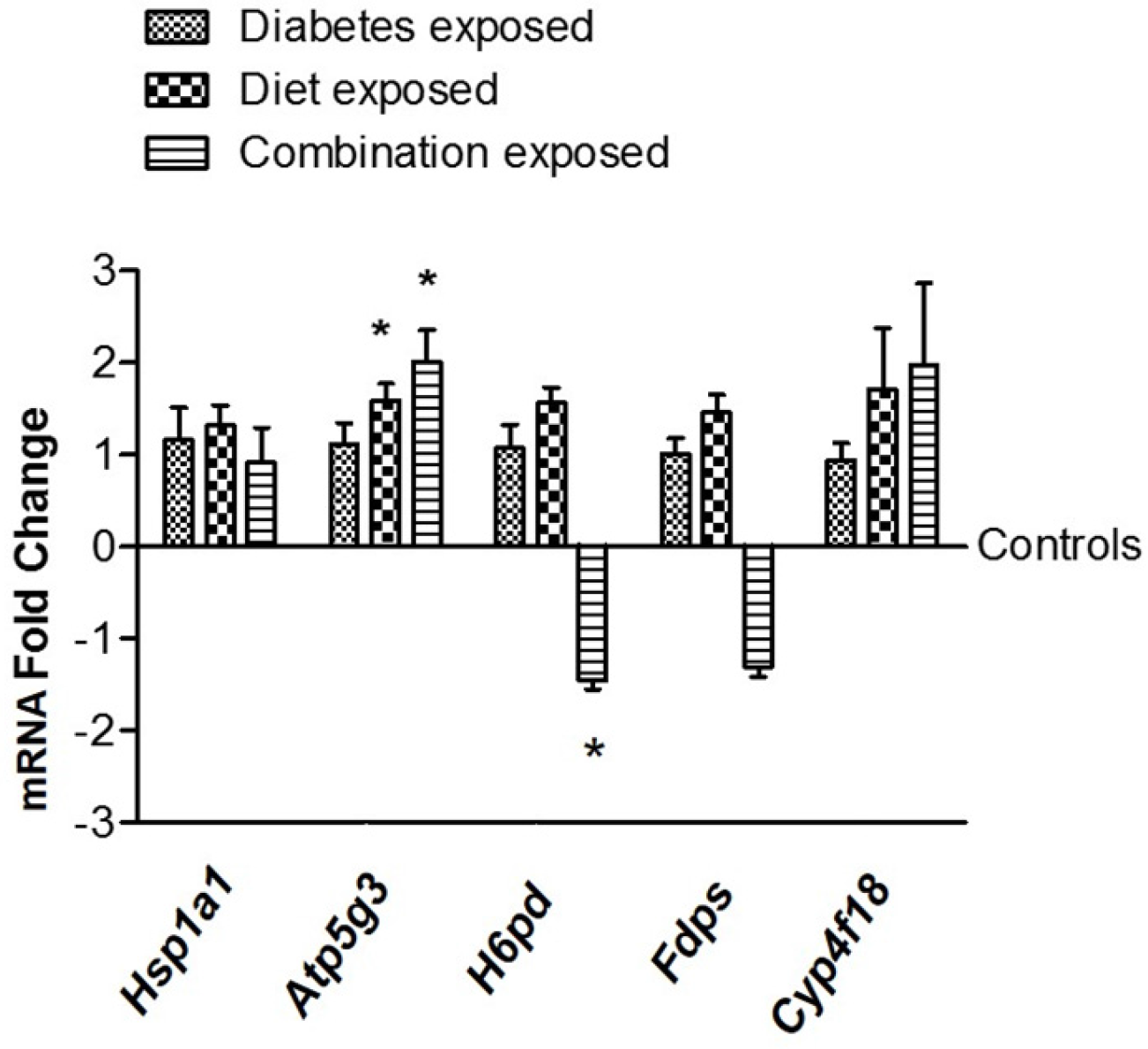
3.6. Gene Expression Validation of Selected Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A



References
- Hod, M.; Kapur, A.; Sacks, D.A.; Hadar, E.; Agarwal, M.; Di Renzo, G.C.; Cabero Roura, L.; McIntyre, H.D.; Morris, J.L.; Divakar, H. The International Federation of Gynecology and Obstetrics (FIGO) Initiative on gestational diabetes mellitus: A pragmatic guide for diagnosis, management, and care. Int. J. Gynaecol. Obstet. 2015, 131, S173–S211. [Google Scholar] [CrossRef]
- Sacks, D.A.; Hadden, D.R.; Maresh, M.; Deerochanawong, C.; Dyer, A.R.; Metzger, B.E.; Lowe, L.P.; Coustan, D.R.; Hod, M.; Oats, J.J.; et al. Frequency of gestational diabetes mellitus at collaborating centers based on IADPSG consensus panel-recommended criteria: The Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Study. Diabetes Care 2012, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Ogden, C.L.; Carroll, M.D.; Fryar, C.D.; Flegal, K.M. Prevalence of obesity among adults and youth: United States, 2011–2014. NCHS Data Brief 2015, 1–8. [Google Scholar]
- Ullmo, S.; Vial, Y.; Di Bernardo, S.; Roth-Kleiner, M.; Mivelaz, Y.; Sekarski, N.; Ruiz, J.; Meijboom, E.J. Pathologic ventricular hypertrophy in the offspring of diabetic mothers: A retrospective study. Eur. Heart J. 2007, 28, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Simeoni, U.; Barker, D.J. Offspring of diabetic pregnancy: Long-term outcomes. Semin. Fetal Neonat. Med. 2009, 14, 119–124. [Google Scholar]
- Eriksson, J.G.; Sandboge, S.; Salonen, M.K.; Kajantie, E.; Osmond, C. Long-term consequences of maternal overweight in pregnancy on offspring later health: Findings from the Helsinki Birth Cohort Study. Ann. Med. 2014, 46, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Perng, W.; Gillman, M.W.; Mantzoros, C.S.; Oken, E. A prospective study of maternal prenatal weight and offspring cardiometabolic health in midchildhood. Ann. Epidemiol. 2014, 24, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Paneni, F.; Cosentino, F. Ageing, metabolism and cardiovascular disease. J. Physiol. 2015, 594, 2061–2073. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Antras, J.; Picatoste, B.; Ramirez, E.; Egido, J.; Tunon, J.; Lorenzo, O. Targeting metabolic disturbance in the diabetic heart. Cardiov. Diabetol. 2015, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- Mdaki, K.S.; Larsen, T.D.; Wachal, A.L.; Schimelpfenig, M.D.; Weaver, L.J.; Dooyema, S.D.; Louwagie, E.J.; Baack, M.L. Maternal high-fat diet impairs cardiac function in offspring of diabetic pregnancy through metabolic stress and mitochondrial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H681–H692. [Google Scholar] [CrossRef] [PubMed]
- Langley-Evans, S.C. Nutritional programming of disease: Unravelling the mechanism. J. Anat. 2009, 215, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Lehnen, H.; Zechner, U.; Haaf, T. Epigenetics of gestational diabetes mellitus and offspring health: The time for action is in early stages of life. Mol. Hum. Reprod. 2013, 19, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Gabory, A.; Attig, L.; Junien, C. Epigenetic mechanisms involved in developmental nutritional programming. World J. Diabetes 2011, 2, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Kathiriya, I.S.; Nora, E.P.; Bruneau, B.G. Investigating the transcriptional control of cardiovascular development. Circ. Res. 2015, 116, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, H.; Rafehi, H.; Bhave, M.; El-Osta, A. Metabolism and chromatin dynamics in health and disease. Mol. Asp. Med. 2016, 54, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Dees, E.; Baldwin, H.S. Making a heart: Advances in understanding the mechanisms of cardiac development. Curr. Opin. Pediatr. 2016, 28, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Jaswal, J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Ashrafian, H.; Frenneaux, M.P.; Opie, L.H. Metabolic mechanisms in heart failure. Circulation 2007, 116, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Mdaki, K.S.; Larsen, T.D.; Weaver, L.J.; Baack, M.L. Age Related Bioenergetics Profiles in Isolated Rat Cardiomyocytes Using Extracellular Flux Analyses. PLoS ONE 2016, 11, e0149002. [Google Scholar] [CrossRef] [PubMed]
- Ong, T.P.; Ozanne, S.E. Developmental programming of type 2 diabetes: Early nutrition and epigenetic mechanisms. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Harmston, N.; Lenhard, B. Chromatin and epigenetic features of long-range gene regulation. Nucleic Acids Res. 2013, 41, 7185–7199. [Google Scholar] [CrossRef] [PubMed]
- Baack, M.L.; Forred, B.J.; Larsen, T.D.; Jensen, D.N.; Wachal, A.L.; Khan, M.A.; Vitiello, P.F. Consequences of a Maternal High-Fat Diet and Late Gestation Diabetes on the Developing Rat Lung. PLoS ONE 2016, 11, e0160818. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Liu, T.; Qin, B.; Zhang, Y.; Liu, X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012, 7, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- FastQC. Available online: http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/ (accessed on 20 April 2016).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.J.; Gazin, C.; Lawson, N.D.; Pages, H.; Lin, S.M.; Lapointe, D.S.; Green, M.R. ChIPpeakAnno: A Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinform. 2010, 11, 237. [Google Scholar] [CrossRef] [PubMed]
- Carroll, T.S.; Liang, Z.; Salama, R.; Stark, R.; de Santiago, I. Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front. Genet. 2014, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.; Krebs, A.R.; Choukrallah, M.A.; Keime, C.; Plewniak, F.; Davidson, I.; Tora, L. seqMINER: An integrated ChIP-seq data interpretation platform. Nucleic Acids Res. 2011, 39, e35. [Google Scholar] [CrossRef] [PubMed]
- Takai, D.; Jones, P.A. The CpG island searcher: A new WWW resource. In Silico Biol. 2003, 3, 235–240. [Google Scholar] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Wang, X.J. GOEAST: A web-based software toolkit for Gene Ontology enrichment analysis. Nucleic Acids Res. 2008, 36, W358–W363. [Google Scholar] [CrossRef] [PubMed]
- Shimoyama, M.; De Pons, J.; Hayman, G.T.; Laulederkind, S.J.; Liu, W.; Nigam, R.; Petri, V.; Smith, J.R.; Tutaj, M.; Wang, S.J.; et al. The Rat Genome Database 2015: Genomic, phenotypic and environmental variations and disease. Nucleic Acids Res. 2015, 43, D743–D750. [Google Scholar] [CrossRef] [PubMed]
- Osborne, J.D.; Flatow, J.; Holko, M.; Lin, S.M.; Kibbe, W.A.; Zhu, L.J.; Danila, M.I.; Feng, G.; Chisholm, R.L. Annotating the human genome with Disease Ontology. BMC Genom. 2009, 10 (Suppl. 1), S6. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Feng, G.; Flatow, J.; Song, J.; Holko, M.; Kibbe, W.A.; Lin, S.M. From disease ontology to disease-ontology lite: Statistical methods to adapt a general-purpose ontology for the test of gene-ontology associations. Bioinformatics 2009, 25, i63–i68. [Google Scholar] [CrossRef] [PubMed]
- Picard. Available online: http://picard.sourceforge.net/ (accessed on 20 April 2016).
- Stark, R.; Brown, G. DiffBind: Differential Binding Analysis of ChIP-Seq Peak Data. Available online: http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf (accessed on 13 June 2016).
- FunDO. Available online: http://fundo.nubic.northwestern.edu (accessed on 20 April 2016).
- Zhu, J.; Adli, M.; Zou, J.Y.; Verstappen, G.; Coyne, M.; Zhang, X.; Durham, T.; Miri, M.; Deshpande, V.; De Jager, P.L.; et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 2013, 152, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Rintisch, C.; Heinig, M.; Bauerfeind, A.; Schafer, S.; Mieth, C.; Patone, G.; Hummel, O.; Chen, W.; Cook, S.; Cuppen, E.; et al. Natural variation of histone modification and its impact on gene expression in the rat genome. Genome Res. 2014, 24, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Thompson, C.B. A two-way street: Reciprocal regulation of metabolism and signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Li, H.S.; Zhang, J.Y.; Thompson, B.S.; Deng, X.Y.; Ford, M.E.; Wood, P.G.; Stolz, D.B.; Eagon, P.K.; Whitcomb, D.C. Rat mitochondrial ATP synthase ATP5G3: Cloning and upregulation in pancreas after chronic ethanol feeding. Physiol. Genom. 2001, 6, 91–98. [Google Scholar]
- Zhao, Y.G.; Zhao, H.; Miao, L.; Wang, L.; Sun, F.; Zhang, H. The p53-induced gene Ei24 is an essential component of the basal autophagy pathway. J. Biol. Chem. 2012, 287, 42053–42063. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, T.; Chen, R.; Wang, X.; Mick, G.; McCormick, K. Effect of diabetes on enzymes involved in rat hepatic corticosterone production. J. Diabetes 2010, 2, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Awazawa, M.; Futami, T.; Sakada, M.; Kaneko, K.; Ohsugi, M.; Nakaya, K.; Terai, A.; Suzuki, R.; Koike, M.; Uchiyama, Y.; et al. Deregulation of pancreas-specific oxidoreductin ERO1beta in the pathogenesis of diabetes mellitus. Mol. Cell. Biol. 2014, 34, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Navarrete, J.M.; Blasco, G.; Xifra, G.; Karczewska-Kupczewska, M.; Stefanowicz, M.; Matulewicz, N.; Puig, J.; Ortega, F.; Ricart, W.; Straczkowski, M.; et al. Obesity Is Associated with Gene Expression and Imaging Markers of Iron Accumulation in Skeletal Muscle. J. Clin. Endocrinol. Metab. 2016, 101, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Vaivoda, R.; Vaine, C.; Boerstler, C.; Galloway, K.; Christmas, P. CYP4F18-Deficient Neutrophils Exhibit Increased Chemotaxis to Complement Component C5a. J. Immunol. Res. 2015, 2015, 250456. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.N.; Czernuszewicz, G.; Tan, Y.; Lombardi, R.; Jin, J.; Willerson, J.T.; Marian, A.J. Human molecular genetic and functional studies identify TRIM63, encoding Muscle RING Finger Protein 1, as a novel gene for human hypertrophic cardiomyopathy. Circ. Res. 2012, 111, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.; Friedman, D.B.; Hill, S.; Caprioli, R.M.; Smith, H.M.; Hill, M.F. Alterations in the diabetic myocardial proteome coupled with increased myocardial oxidative stress underlies diabetic cardiomyopathy. J. Mol. Cell. Cardiol. 2007, 42, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.T.; Harris, R.A.; French, S.; Blair, P.V.; You, J.; Bemis, K.G.; Wang, M.; Balaban, R.S. Tissue heterogeneity of the mammalian mitochondrial proteome. Am. J. Physiol. Cell. Physiol. 2007, 292, C689–C697. [Google Scholar] [CrossRef] [PubMed]
- Ares-Carrasco, S.; Picatoste, B.; Camafeita, E.; Carrasco-Navarro, S.; Zubiri, I.; Ortiz, A.; Egido, J.; Lopez, J.A.; Tunon, J.; Lorenzo, O. Proteome changes in the myocardium of experimental chronic diabetes and hypertension: Role of PPARalpha in the associated hypertrophy. J. Proteomics 2012, 75, 1816–1829. [Google Scholar] [CrossRef] [PubMed]
- Jenei, Z.M.; Gombos, T.; Forhecz, Z.; Pozsonyi, Z.; Karadi, I.; Janoskuti, L.; Prohaszka, Z. Elevated extracellular HSP70 (HSPA1A) level as an independent prognostic marker of mortality in patients with heart failure. Cell Stress Chaperones 2013, 18, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Camargo, A.; Rangel-Zuniga, O.A.; Pena-Orihuela, P.; Marin, C.; Perez-Martinez, P.; Delgado-Lista, J.; Gutierrez-Mariscal, F.M.; Malagon, M.M.; Roche, H.M.; Tinahones, F.J.; et al. Postprandial changes in the proteome are modulated by dietary fat in patients with metabolic syndrome. J. Nutr. Biochem. 2013, 24, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Garamvolgyi, Z.; Prohaszka, Z.; Rigo, J., Jr.; Kecskemeti, A.; Molvarec, A. Increased circulating heat shock protein 70 (HSPA1A) levels in gestational diabetes mellitus: A pilot study. Cell Stress Chaperones 2015, 20, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Jenei, Z.M.; Szeplaki, G.; Merkely, B.; Karadi, I.; Zima, E.; Prohaszka, Z. Persistently elevated extracellular HSP70 (HSPA1A) level as an independent prognostic marker in post-cardiac-arrest patients. Cell Stress Chaperones 2013, 18, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Riddick, D.S.; Ding, X.; Wolf, C.R.; Porter, T.D.; Pandey, A.V.; Zhang, Q.Y.; Gu, J.; Finn, R.D.; Ronseaux, S.; McLaughlin, L.A.; et al. NADPH-cytochrome P450 oxidoreductase: Roles in physiology, pharmacology, and toxicology. Drug Metab. Dispos. 2013, 41, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, X.; Chen, L.; Wu, J.; Dang, H.; Wei, M.; Fan, Y.; Zhang, Y.; Zhu, Y.; Wang, N.; et al. Expression profiling of hepatic genes associated with lipid metabolism in nephrotic rats. Am. J. Physiol. Renal Physiol. 2008, 295, F662–F671. [Google Scholar] [CrossRef] [PubMed]
- Harder, M.N.; Appel, E.V.; Grarup, N.; Gjesing, A.P.; Ahluwalia, T.S.; Jorgensen, T.; Christensen, C.; Brandslund, I.; Linneberg, A.; Sorensen, T.I.; et al. The type 2 diabetes risk allele of TMEM154-rs6813195 associates with decreased beta cell function in a study of 6486 Danes. PLoS ONE 2015, 10, e0120890. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; He, J.; Kucera, H.; Gaikwad, N.W.; Zhang, B.; Xu, M.; O’Doherty, R.M.; Selcer, K.W.; Xie, W. Hepatic overexpression of steroid sulfatase ameliorates mouse models of obesity and type 2 diabetes through sex-specific mechanisms. J. Biol. Chem. 2014, 289, 8086–8097. [Google Scholar] [CrossRef] [PubMed]
- Liew, O.W.; Yandle, T.G.; Chong, J.P.; Ng, Y.X.; Frampton, C.M.; Ng, T.P.; Lam, C.S.; Richards, A.M. High-Sensitivity Sandwich ELISA for Plasma NT-proUcn2: Plasma Concentrations and Relationship to Mortality in Heart Failure. Clin. Chem. 2016, 62, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Noma, T.; Fujisawa, K.; Yamashiro, Y.; Shinohara, M.; Nakazawa, A.; Gondo, T.; Ishihara, T.; Yoshinobu, K. Structure and expression of human mitochondrial adenylate kinase targeted to the mitochondrial matrix. Biochem. J. 2001, 358, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Zhang, Y.; Liu, Y.; Chen, J.; Zong, C.; Yu, C.; Cui, S.; Gao, W.; Qin, D.; Sun, W.; et al. Signal transducer and activator of transcription 5B (STAT5B) modulates adipocyte differentiation via MOF. Cell Signal. 2015, 27, 2434–2443. [Google Scholar] [CrossRef] [PubMed]
- Seet, E.L.; Yee, J.K.; Jellyman, J.K.; Han, G.; Ross, M.G.; Desai, M. Maternal high-fat-diet programs rat offspring liver fatty acid metabolism. Lipids 2015, 50, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Berezin, A. Epigenetics in heart failure phenotypes. BBA Clin. 2016, 6, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Sack, M.N. Type 2 diabetes, mitochondrial biology and the heart. J. Mol. Cell. Cardiol. 2009, 46, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Wang, H.; Cui, H.; Chen, H.; Pan, Y.X. Early-life exposure to high-fat diet may predispose rats to gender-specific hepatic fat accumulation by programming Pepck expression. J. Nutr. Biochem. 2015, 26, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.F.; Cai, W.; Xu, J.L.; Shi, W. Maternal high-fat diet programs Wnt genes through histone modification in the liver of neonatal rats. J. Mol. Endocrinol. 2012, 49, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Borengasser, S.J.; Kang, P.; Faske, J.; Gomez-Acevedo, H.; Blackburn, M.L.; Badger, T.M.; Shankar, K. High fat diet and in utero exposure to maternal obesity disrupts circadian rhythm and leads to metabolic programming of liver in rat offspring. PLoS ONE 2014, 9, e84209. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Histone Modification | CC vs. CS (Gained/Lost Peaks) | CC vs. HC (Gained/Lost Peaks) | CS vs. HS (Gained/Lost Peaks) | |||
|---|---|---|---|---|---|---|
| All Region | Promoter Region | All Region | Promoter Region | All Region | Promoter Region | |
| H3Ac | 0/32 | 0/2 | 5/103 | 0/3 | 0/0 | 0/0 |
| H3K4me3 | 8/6 | 2/0 | 449/11 | 28/0 | 1/6 | 1/0 |
| H3K27me3 | 186/82 | 6/3 | 309/114 | 6/1 | 403/226 | 4/22 |
| Modification | Diets | Genes | Gene Names | Fold Change | FDR |
|---|---|---|---|---|---|
| H3Ac | CC vs. CS | Myrf | Myelin regulatory factor | −5.53 | 1.59 × 10−2 |
| H3K4me3 | CC vs. CS | Hspa1a | Heat shock protein 1A | 6.54 | 2.49 × 10−2 |
| Hspa1b | Heat shock protein 1B | 6.54 | 2.49 × 10−2 | ||
| CC vs. HC | Atp5g3 | ATP synthase, subunit C3 | 6.10 | 7.63 × 10−2 | |
| Cyp4f18 | Cytochrome P450, polypeptide 18 | 5.06 | 1.27 × 10−2 | ||
| Ei24 | EI24, autophagy associated transmembrane protein | 2.79 | 2.42 × 10−2 | ||
| Ero1lb | Endoplasmic reticulum oxidoreductase beta | 6.66 | 1.96 × 10−2 | ||
| Fdps | Farnesyl diphosphate synthase | 1.92 | 5.66 × 10−2 | ||
| H6pd | Hexose-6-phosphate dehydrogenase | 6.75 | 7.39 × 10−2 | ||
| Hspa1a | Heat shock protein 1A | 5.62 | 3.76 × 10−2 | ||
| Hspa1b | Heat shock protein 1B | 5.62 | 3.76 × 10−2 | ||
| Mapk8ip2 | Mitogen-activated protein kinase 8 interacting protein 2 | 6.16 | 6.08 × 10−2 | ||
| Napsa | Napsin A aspartic peptidase | 6.18 | 5.66 × 10−2 | ||
| Nfix | Nuclear factor I/X (CCAAT-binding transcription factor) | 2.09 | 8.40 × 10−2 | ||
| Por | P450 (cytochrome) oxidoreductase | 6.16 | 6.08 × 10−2 | ||
| Slc11a2 | Solute carrier family 11, member 2 | 6.73 | 9.55 × 10−2 | ||
| Trim63 | Tripartite motif containing 63 | 5.00 | 9.51 × 10−2 | ||
| Zim1 | Zinc finger, imprinted 1 | 6.28 | 3.43 × 10−2 | ||
| H3K27me3 | CC vs. CS | Bmp8b | Bone morphogenetic protein 8b | 2.25 | 3.03 × 10−2 |
| Gli1 | GLI family zinc finger 1 | 2.77 | 6.51 × 10−2 | ||
| Rps29 | Ribosomal protein S29 | 3.73 | 7.29 × 10−2 | ||
| Snapc3 | Small nuclear RNA activating complex, polypeptide 3 | 4.51 | 7.29 × 10−2 | ||
| Ccdc126 | Coiled-coil domain containing 126 | −4.10 | 9.17 × 10−2 | ||
| CC vs. HC | Dstyk | Dual serine/threonine and tyrosine protein kinase | 2.57 | 8.56 × 10−2 | |
| Lhpp | Phospholysine phosphohistidine inorganic pyrophosphate phosphatase | 5.63 | 1.69 × 10−2 | ||
| Zfp444 | Zinc finger protein 668 | −5.83 | 4.77 × 10−2 | ||
| CS vs. HS | Ak3 | Adenylate kinase 3 | 1.95 | 9.12 × 10−2 | |
| Stat5b | Signal transducer and activator of transcription 5B | 3.76 | 8.66 × 10−2 | ||
| Abcb9 | ATP binding cassette subfamily B member 9 | −5.60 | 2.22 × 10−2 | ||
| Atp5g2 | ATP synthase, mitochondrial Fo complex, subunit C2 | −5.92 | 3.41 × 10−2 | ||
| B3gnt3 | UDP-glcnac:betagal beta-1,3-N-acetylglucosaminyltransferase 3 | −5.79 | 7.71 × 10−2 | ||
| Kdm6b | Lysine demethylase 6B | −5.37 | 7.80 × 10−2 | ||
| Ldb1 | LIM domain binding 1 | −5.73 | 1.10 × 10−2 | ||
| Metap1 | Methionyl aminopeptidase 1 | −5.58 | 2.46 × 10−2 | ||
| Nfic | Nuclear factor I/C | −4.58 | 3.65 × 10−2 | ||
| Nr4a3 | Nuclear receptor subfamily 4, group A, member 3 | −3.41 | 3.70 × 10−2 | ||
| Ruvbl2 | Ruvb-like AAA atpase 2 | −3.40 | 5.43 × 10−2 | ||
| Slc44a4 | Solute carrier family 44, member 4 | −6.18 | 2.79 × 10−2 | ||
| Snapc3 | Small nuclear RNA activating complex, polypeptide 3 | −4.17 | 4.90 × 10−2 | ||
| Sts | Steroid sulfatase (microsomal), isozyme S | −1.82 | 7.95 × 10−2 | ||
| Ucn2 | Urocortin 2 | −2.61 | 5.70 × 10−2 | ||
| Uqcrq | Ubiquinol-cytochrome c reductase, complex III subunit VII | −5.73 | 1.10 × 10−2 |
| Gene | Enriched GO ID | Description of GO | Log 2 Odd Ratio | FDR |
|---|---|---|---|---|
| Atp5g3 | GO:1901360 | Organic cyclic compound metabolic process | 1.96 | 0.0976 |
| GO:0044238 | Primary metabolic process | 1.38 | 0.0636 | |
| Cyp4f18 | GO:1901360 | Organic cyclic compound metabolic process | 1.96 | 0.0976 |
| GO:0044248 | Cellular catabolic process | 2.87 | 0.0976 | |
| GO:0044238 | Primary metabolic process | 1.38 | 0.0636 | |
| GO:0016709 | Oxidoreductase activity | 6.13 | 0.0976 | |
| Ei24 | GO:0030308 | Negative regulation of cell growth | 4.80 | 0.0726 |
| Fdps | GO:0045542 | Positive regulation of cholesterol biosynthetic process | 8.68 | 0.0195 |
| GO:1901360 | Organic cyclic compound metabolic process | 1.96 | 0.0976 | |
| GO:0044238 | Primary metabolic process | 1.38 | 0.0636 | |
| H6pd | GO:1901360 | Organic cyclic compound metabolic process | 1.96 | 0.0976 |
| GO:0044238 | Primary metabolic process | 1.38 | 0.0636 | |
| Hspa1a | GO:0008180 | COP9 signalosome | 6.31 | 0.0976 |
| GO:1901360 | Organic cyclic compound metabolic process | 1.96 | 0.0976 | |
| GO:0044248 | Cellular catabolic process | 2.87 | 0.0976 | |
| GO:0090083 | Regulation of inclusion body assembly | 8.09 | 0.0312 | |
| GO:0044238 | Primary metabolic process | 1.38 | 0.0636 | |
| Hspa1b | GO:0008180 | COP9 signalosome | 6.31 | 0.0976 |
| GO:0043281 | Regulation of cysteine-type endopeptidase activity involved in apoptotic process | 4.43 | 0.0976 | |
| GO:1901360 | Organic cyclic compound metabolic process | 1.96 | 0.0976 | |
| GO:0044248 | Cellular catabolic process | 2.87 | 0.0976 | |
| GO:0090083 | Regulation of inclusion body assembly | 8.09 | 0.0312 | |
| GO:0044238 | Primary metabolic process | 1.38 | 0.0636 | |
| Mapk8ip2 | GO:0044238 | Primary metabolic process | 1.38 | 0.0636 |
| Napsa | GO:0044238 | Primary metabolic process | 1.38 | 0.0636 |
| Nfix | GO:1901360 | Organic cyclic compound metabolic process | 1.96 | 0.0976 |
| GO:0044238 | Primary metabolic process | 1.38 | 0.0636 | |
| Por | GO:0045542 | Positive regulation of cholesterol biosynthetic process | 8.68 | 0.0195 |
| GO:0043281 | Regulation of cysteine-type endopeptidase activity involved in apoptotic process | 4.43 | 0.0976 | |
| GO:0044248 | Cellular catabolic process | 2.87 | 0.0976 | |
| GO:0044238 | Primary metabolic process | 1.38 | 0.0636 | |
| GO:0016709 | Oxidoreductase activity | 6.13 | 0.0976 | |
| Slc11a2 | GO:0043281 | Regulation of cysteine-type endopeptidase activity | 4.43 | 0.0976 |
| GO:1901360 | Organic cyclic compound metabolic process | 1.96 | 0.0976 | |
| GO:0044238 | Primary metabolic process | 1.38 | 0.0636 | |
| Trim63 | GO:0044248 | Cellular catabolic process | 2.87 | 0.0976 |
| GO:0044238 | Primary metabolic process | 1.38 | 0.0636 |
| Gene | QTL | QTL Count (Percent Total) | ||||||
|---|---|---|---|---|---|---|---|---|
| Blood Pressure | Body Weight | Glucose Level | Insulin Level | Insulin Dependent Diabetes Mellitus | Non-insulin Dependent Diabetes Mellitus | Serum Cholesterol | ||
| Atp5g3 | 22 | 7 (31.82%) | 9 (40.91%) | 1 (4.55%) | 1 (4.55%) | |||
| Cyp4f18 | 14 | 3 (21.43% | 3 (21.43%) | 2 (14.29%) | 1 (7.14%) | 1 (7.14%) | ||
| Ei24 | 19 | 7 (36.84%) | 2 (10.53%) | 1 (5.26%) | 2 (10.53%) | 2 (10.53%) | ||
| Ero1lb | 19 | 7 (36.84%) | 1 (5.26%) | 2 (10.53%) | 1 (5.26%) | 2 (10.53%) | 3 (15.79%) | |
| Fdps | 33 | 22 (66.67%) | 3 (9.09%) | 2 (6.06%) | ||||
| H6pd | 12 | 7 (58.33%) | 2 (16.67%) | |||||
| Hspa1a | 3 | 1 (33.33%) | 1 (33.33%) | 1 (33.33%) | ||||
| Hspa1b | 3 | 1 (33.33%) | 1 (33.33%) | 1 (33.33%) | ||||
| Mapk8ip2 | 12 | 6 (50.00%) | 1 (8.33%) | 2 (16.67%) | 1 (8.33%) | |||
| Napsa | 16 | 7 (43.75%) | 1 (6.25%) | 1 (6.25%) | 2 (12.50%) | 2 (12.50%) | ||
| Nfix | 4 | 1 (25.00%) | 1 (25.00%) | |||||
| Por | 12 | 6 (50.00%) | 1 (8.33%) | 1 (8.33%) | 1 (8.33%) | 1 (8.33%) | 2 (16.67%) | |
| Slc11a2 | 8 | 5 (62.50%) | 1 (12.50%) | |||||
| Trim63 | 19 | 7 (36.84%) | 3 (15.79%) | 1 (5.26%) | 4 (21.05%) | |||
| Zim1 | 6 | 1 (16.67%) | 2 (33.33%) | 1 (16.67%) | 1 (16.67%) | |||
| Total | 455 | 88 (19.34%) | 24 (5.27%) | 7 (1.54%) | 5 (1.10%) | 4 (0.88%) | 15 (3.30%) | 20 (4.40%) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Upadhyaya, B.; Larsen, T.; Barwari, S.; Louwagie, E.J.; Baack, M.L.; Dey, M. Prenatal Exposure to a Maternal High-Fat Diet Affects Histone Modification of Cardiometabolic Genes in Newborn Rats. Nutrients 2017, 9, 407. https://doi.org/10.3390/nu9040407
Upadhyaya B, Larsen T, Barwari S, Louwagie EJ, Baack ML, Dey M. Prenatal Exposure to a Maternal High-Fat Diet Affects Histone Modification of Cardiometabolic Genes in Newborn Rats. Nutrients. 2017; 9(4):407. https://doi.org/10.3390/nu9040407
Chicago/Turabian StyleUpadhyaya, Bijaya, Tricia Larsen, Shivon Barwari, Eli J. Louwagie, Michelle L. Baack, and Moul Dey. 2017. "Prenatal Exposure to a Maternal High-Fat Diet Affects Histone Modification of Cardiometabolic Genes in Newborn Rats" Nutrients 9, no. 4: 407. https://doi.org/10.3390/nu9040407
APA StyleUpadhyaya, B., Larsen, T., Barwari, S., Louwagie, E. J., Baack, M. L., & Dey, M. (2017). Prenatal Exposure to a Maternal High-Fat Diet Affects Histone Modification of Cardiometabolic Genes in Newborn Rats. Nutrients, 9(4), 407. https://doi.org/10.3390/nu9040407