Fructose, Glucocorticoids and Adipose Tissue: Implications for the Metabolic Syndrome

 ,
,  ,
, 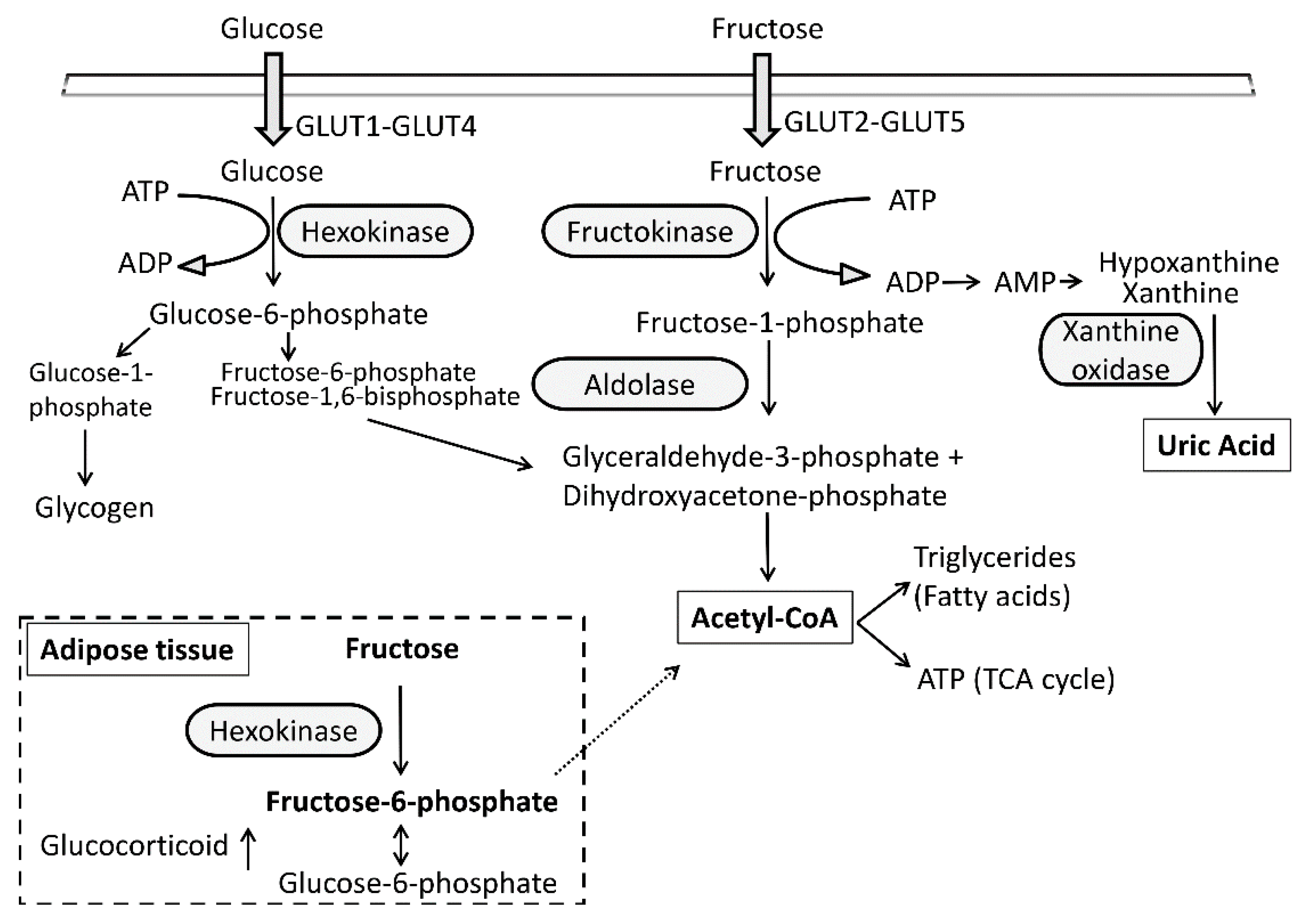{kind=link}
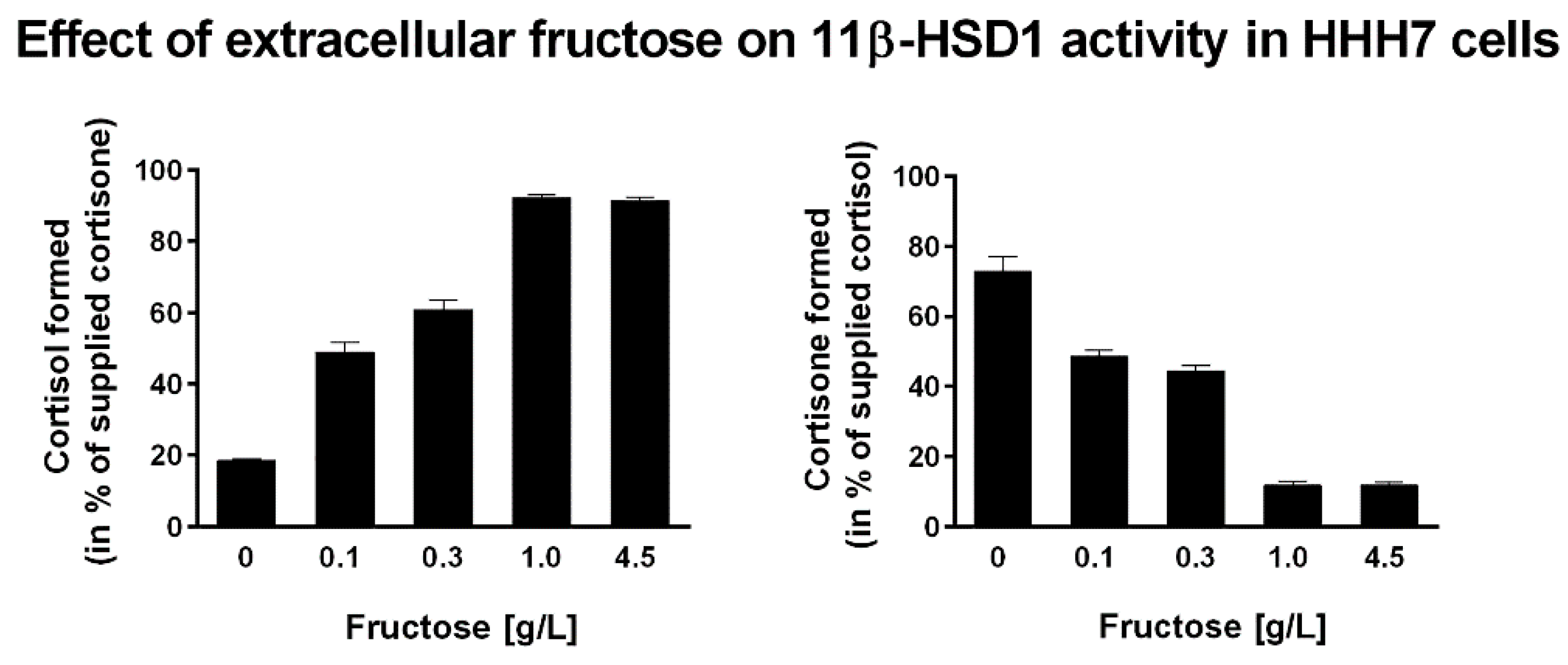{kind=link}
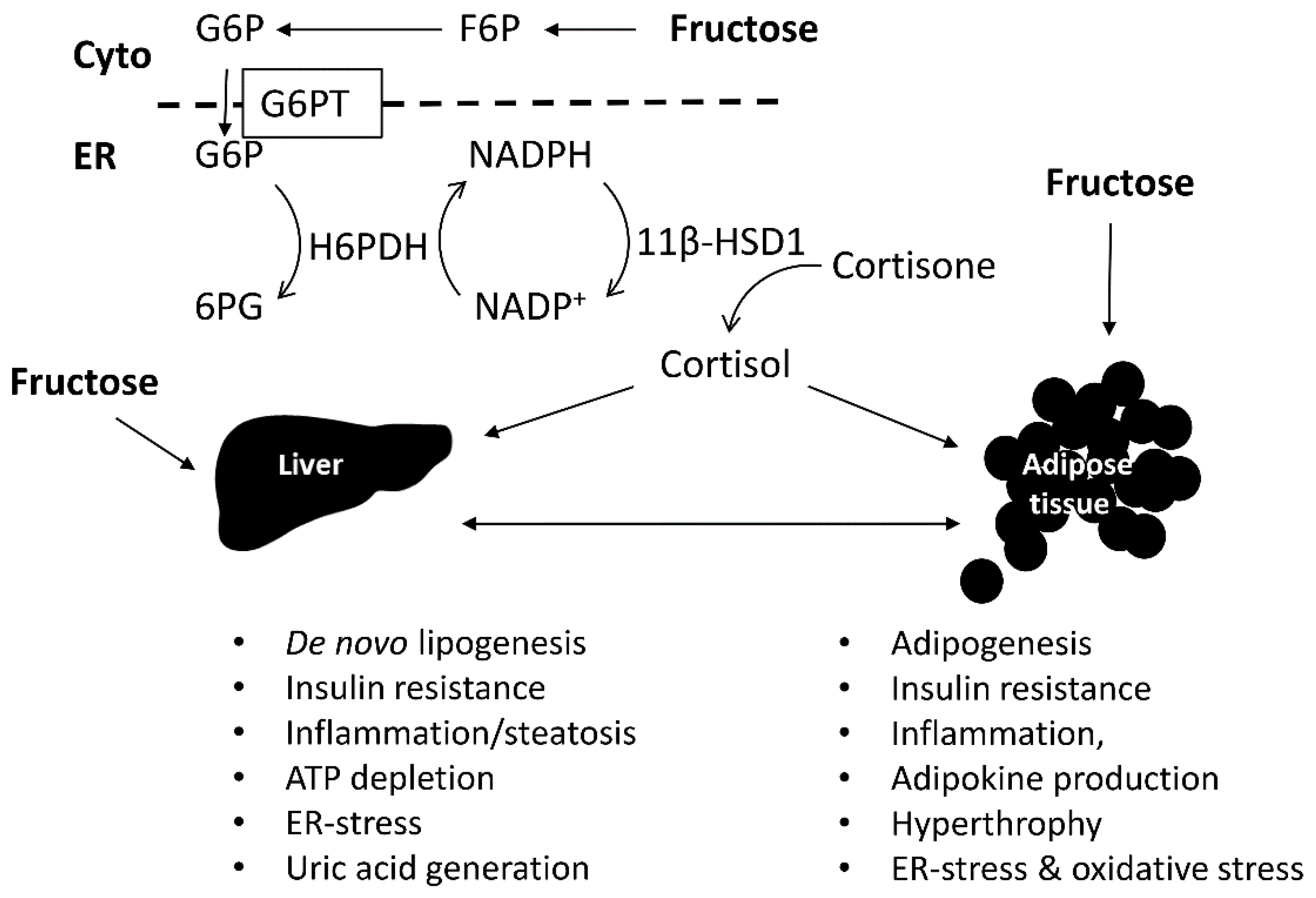{kind=link}
Abstract
:1. Introduction
2. Dietary Fructose and Adiposity
3. Fructose Metabolism in Adipose Tissue
4. Effect of Fructose on Metabolic Disturbances
5. Role of 11β-HSD1 in Adipocyte Differentiation/Proliferation
6. Effect of Fructose on 11β-HSD1 Expression and Activity
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Malik, V.S.; Hu, F.B. Fructose and Cardiometabolic Health: What the Evidence from Sugar-Sweetened Beverages Tells Us. J. Am. Coll. Cardiol. 2015, 66, 1615–1624. [Google Scholar] [CrossRef] [PubMed]
- Ogden, C.L.; Carroll, M.D.; Curtin, L.R.; McDowell, M.A.; Tabak, C.J.; Flegal, M.K. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA 2006, 295, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.S.; Popkin, B.M.; Bray, G.A.; Despres, J.P.; Hu, F.B. Sugar-sweetened beverages, obesity, type 2 diabetes mellitus, and cardiovascular disease risk. Circulation 2010, 121, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.S.; Popkin, B.M.; Bray, G.A.; Despres, J.P.; Willett, W.C.; Hu, F.B. Sugar-sweetened beverages and risk of metabolic syndrome and type 2 diabetes: A meta-analysis. Diabetes Care 2010, 33, 2477–2483. [Google Scholar] [CrossRef] [PubMed]
- Yudkin, J.; Roddy, J. Levels of Dietary Sucrose in Patients with Occlusive Atherosclerotic Disease. Lancet 1964, 2, 6–8. [Google Scholar] [CrossRef]
- Bantle, J.P.; Laine, D.C.; Thomas, J.W. Metabolic effects of dietary fructose and sucrose in types I and II diabetic subjects. JAMA 1986, 256, 3241–3246. [Google Scholar] [CrossRef] [PubMed]
- Despres, J.P.; Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 2006, 444, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Cristancho, A.G.; Lazar, M.A. Forming functional fat: A growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 2011, 12, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol. Rev. 2013, 93, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C.; Ahluwalia, N.; Brouns, F.; Buetler, T.; Clement, K.; Cunningham, K.; Esposito, K.; Jonsson, L.S.; Kolb, H.; Lansink, M.; et al. Dietary factors and low-grade inflammation in relation to overweight and obesity. Br. J. Nutr. 2011, 106 (Suppl. 3), S5–S78. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.K.; Yetley, E.A. Intakes and food sources of fructose in the United States. Am. J. Clin. Nutr. 1993, 58 (Suppl. 5), 737S–747S. [Google Scholar] [PubMed]
- Vos, M.B.; Kimmons, J.E.; Gillespie, C.; Welsh, J.; Blanck, H.M. Dietary fructose consumption among US children and adults: The Third National Health and Nutrition Examination Survey. Medscape J. Med. 2008, 10, 160. [Google Scholar] [PubMed]
- Bray, G.A.; Nielsen, S.J.; Popkin, B.M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004, 79, 537–543. [Google Scholar] [PubMed]
- Stanhope, K.L.; Havel, P.J. Fructose consumption: Potential mechanisms for its effects to increase visceral adiposity and induce dyslipidemia and insulin resistance. Curr. Opin. Lipidol. 2008, 19, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Le, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef] [PubMed]
- Bizeau, M.E.; Pagliassotti, M.J. Hepatic adaptations to sucrose and fructose. Metabolism 2005, 54, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Hanover, L.M.; White, J.S. Manufacturing, composition, and applications of fructose. Am. J. Clin. Nutr. 1993, 58 (Suppl. 5), 724S–732S. [Google Scholar] [PubMed]
- Zubiria, M.G.; Alzamendi, A.; Moreno, G.; Rey, M.A.; Spinedi, E.; Giovambattista, A. Long-Term Fructose Intake Increases Adipogenic Potential: Evidence of Direct Effects of Fructose on Adipocyte Precursor Cells. Nutrients 2016, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Masuzaki, H.; Paterson, J.; Shinyama, H.; Morton, N.M.; Mullins, J.J.; Seckl, J.R.; Flier, J.S. A transgenic model of visceral obesity and the metabolic syndrome. Science 2001, 294, 2166–2170. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.W.; Finney, J.; Gay, C.; Hughes, B.A.; Hughes, S.V.; Stewart, P.M. Impaired glucose tolerance and insulin resistance are associated with increased adipose 11beta-hydroxysteroid dehydrogenase type 1 expression and elevated hepatic 5alpha-reductase activity. Diabetes 2008, 57, 2652–2660. [Google Scholar] [CrossRef] [PubMed]
- Morton, N.M. Obesity and corticosteroids: 11beta-hydroxysteroid type 1 as a cause and therapeutic target in metabolic disease. Mol. Cell Endocrinol. 2010, 316, 154–164. [Google Scholar] [CrossRef] [PubMed]
- London, E.; Castonguay, T.W. High fructose diets increase 11beta-hydroxysteroid dehydrogenase type 1 in liver and visceral adipose in rats within 24-h exposure. Obesity 2011, 19, 925–932. [Google Scholar] [CrossRef] [PubMed]
- McCormick, K.L.; Wang, X.; Mick, G.J. Modification of microsomal 11beta-HSD1 activity by cytosolic compounds: Glutathione and hexose phosphoesters. J. Steroid. Biochem. Mol. Biol. 2008, 111, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Senesi, S.; Legeza, B.; Balazs, Z.; Csala, M.; Marcolongo, P.; Kereszturi, E.; Szelenyi, P.; Egger, C.; Fulceri, R.; Mandl, J.; et al. Contribution of fructose-6-phosphate to glucocorticoid activation in the endoplasmic reticulum: Possible implication in the metabolic syndrome. Endocrinology 2010, 151, 4830–4839. [Google Scholar] [CrossRef] [PubMed]
- Staab, C.A.; Maser, E. 11beta-Hydroxysteroid dehydrogenase type 1 is an important regulator at the interface of obesity and inflammation. J. Steroid Biochem. Mol. Biol. 2010, 119, 56–72. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Pramyothin, P.; Karastergiou, K.; Fried, S.K. Deconstructing the roles of glucocorticoids in adipose tissue biology and the development of central obesity. Biochim. Biophys. Acta 2014, 1842, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Banhegyi, G.; Benedetti, A.; Fulceri, R.; Senesi, S. Cooperativity between 11beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase in the lumen of the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 27017–27021. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Nashev, L.G.; Schweizer, R.A.; Frick, C.; Odermatt, A. Hexose-6-phosphate dehydrogenase determines the reaction direction of 11beta-hydroxysteroid dehydrogenase type 1 as an oxoreductase. FEBS Lett. 2004, 571, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Odermatt, A.; Arnold, P.; Stauffer, A.; Frey, B.M.; Frey, F.J. The N-terminal anchor sequences of 11beta-hydroxysteroid dehydrogenases determine their orientation in the endoplasmic reticulum membrane. J. Biol. Chem. 1999, 274, 28762–28770. [Google Scholar] [CrossRef] [PubMed]
- Ozols, J. Lumenal orientation and post-translational modifications of the liver microsomal 11 beta-hydroxysteroid dehydrogenase. J. Biol. Chem. 1995, 270, 2305–2312. [Google Scholar] [PubMed]
- Odermatt, A.; Kratschmar, D.V. Tissue-specific modulation of mineralocorticoid receptor function by 11beta-hydroxysteroid dehydrogenases: An overview. Mol. Cell Endocrinol. 2012, 350, 168–186. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.W.; Walker, E.A.; Bujalska, I.J.; Draper, N.; Lavery, G.G.; Cooper, M.S.; Hewison, M.; Stewart, P.M. 11beta-hydroxysteroid dehydrogenase type 1: A tissue-specific regulator of glucocorticoid response. Endocr. Rev. 2004, 25, 831–866. [Google Scholar] [CrossRef] [PubMed]
- Seckl, J.R. 11beta-hydroxysteroid dehydrogenases: Changing glucocorticoid action. Curr. Opin. Pharmacol. 2004, 4, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Legeza, B.; Balazs, Z.; Odermatt, A. Fructose promotes the differentiation of 3T3-L1 adipocytes and accelerates lipid metabolism. FEBS Lett. 2014, 588, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Sharma, A.; Abramson, J.L.; Vaccarino, V.; Gillespie, C.; Vos, M.B. Caloric sweetener consumption and dyslipidemia among US adults. JAMA 2010, 303, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Yoffe, P.; Hills, N.; Lustig, R.H. The relationship of sugar to population-level diabetes prevalence: An econometric analysis of repeated cross-sectional data. PLoS ONE 2013, 8, e57873. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.S.; Keim, N.L.; Stern, J.S.; Teff, K.; Havel, P.J. Fructose, weight gain, and the insulin resistance syndrome. Am. J. Clin. Nutr. 2002, 76, 911–922. [Google Scholar] [PubMed]
- Johnson, R.J.; Segal, M.S.; Sautin, Y.; Nakagawa, T.; Feig, D.I.; Kang, D.H.; Gersch, M.S.; Benner, S.; Sanchez-Lozada, L.G. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am. J. Clin. Nutr. 2007, 86, 899–906. [Google Scholar] [PubMed]
- Dennison, B.A.; Rockwell, H.L.; Baker, S.L. Excess fruit juice consumption by preschool-aged children is associated with short stature and obesity. Pediatrics 1997, 99, 15–22. [Google Scholar] [PubMed]
- Ludwig, D.S.; Peterson, K.E.; Gortmaker, S.L. Relation between consumption of sugar-sweetened drinks and childhood obesity: A prospective, observational analysis. Lancet 2001, 357, 505–508. [Google Scholar] [CrossRef]
- Schulze, M.B.; Manson, J.E.; Ludwig, D.S.; Colditz, G.A.; Stampfer, M.J.; Willett, W.C.; Hu, F.B. Sugar-sweetened beverages, weight gain, and incidence of type 2 diabetes in young and middle-aged women. JAMA 2004, 292, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; McKeown, N.M.; Hwang, S.J.; Hoffmann, U.; Jacques, P.F.C.S. Fox, Sugar-Sweetened Beverage Consumption Is Associated With Change of Visceral Adipose Tissue Over 6 Years of Follow-Up. Circulation 2016, 133, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Ha, V.; Jayalath, V.H.; Cozma, A.I.; Mirrahimi, A.; de Souza, R.J.; Sievenpiper, J.L. Fructose-containing sugars, blood pressure, and cardiometabolic risk: A critical review. Curr. Hypertens. Rep. 2013, 15, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Sievenpiper, J.L.; de Souza, R.J.; Mirrahimi, A.; Yu, M.E.; Carleton, A.J.; Beyene, J.; Chiavaroli, L.; di Buono, M.; Jenkins, A.L.; Leiter, L.A.; et al. Effect of fructose on body weight in controlled feeding trials: A systematic review and meta-analysis. Ann Intern. Med. 2012, 156, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Sievenpiper, J.L.; de Souza, R.J.; Chiavaroli, L.; Ha, V.; Cozma, A.I.; Mirrahimi, A.; Yu, M.E.; Carleton, A.J.; di Buono, M.; et al. The effects of fructose intake on serum uric acid vary among controlled dietary trials. J. Nutr. 2012, 142, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaka, T.; Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Ohashi, K.; Tomita, S.; Sekiya, M.; et al. Insulin-independent induction of sterol regulatory element-binding protein-1c expression in the livers of streptozotocin-treated mice. Diabetes 2004, 53, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Bantle, J.P.; Raatz, S.K.; Thomas, W.; Georgopoulos, A. Effects of dietary fructose on plasma lipids in healthy subjects. Am. J. Clin. Nutr. 2000, 72, 1128–1134. [Google Scholar] [PubMed]
- Bantle, J.P.; Swanson, J.E.; Thomas, W.; Laine, D.C. Metabolic effects of dietary fructose in diabetic subjects. Diabetes Care 1992, 15, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.; Mu, W.; Roncal, C.; Cheng, K.Y.; Johnson, R.J.; Scarpace, P.J. Fructose-induced leptin resistance exacerbates weight gain in response to subsequent high-fat feeding. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1370–R1375. [Google Scholar] [CrossRef] [PubMed]
- Le, K.A.; Faeh, D.; Stettler, R.; Ith, M.; Kreis, R.; Vermathen, P.; Boesch, C.; Ravussin, E.; Tappy, L. A 4-wk high-fructose diet alters lipid metabolism without affecting insulin sensitivity or ectopic lipids in healthy humans. Am. J. Clin. Nutr. 2006, 84, 1374–1379. [Google Scholar] [PubMed]
- Le, K.A.; Ith, M.; Kreis, R.; Faeh, D.; Bortolotti, M.; Tran, C.; Boesch, C.; Tappy, L. Fructose overconsumption causes dyslipidemia and ectopic lipid deposition in healthy subjects with and without a family history of type 2 diabetes. Am. J. Clin. Nutr. 2009, 89, 1760–1765. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Hu, H.; Zharikov, S.; Tuttle, K.R.; Short, R.A.; Glushakova, O.; Ouyang, X.; Feig, D.I.; Block, E.R.; Herrera-Acosta, J.; et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am. J. Physiol. Renal. Physiol. 2006, 290, F625–F631. [Google Scholar] [CrossRef] [PubMed]
- Bremer, A.A.; Stanhope, K.L.; Graham, J.L.; Cummings, B.P.; Wang, W.; Saville, B.R.; Havel, P.J. Fructose-fed rhesus monkeys: A nonhuman primate model of insulin resistance, metabolic syndrome, and type 2 diabetes. Clin. Transl. Sci. 2011, 4, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Perez-Pozo, S.E.; Sautin, Y.Y.; Manitius, J.; Sanchez-Lozada, L.G.; Feig, D.I.; Shafiu, M.; Segal, M.; Glassock, R.J.; Shimada, M.; et al. Hypothesis: Could excessive fructose intake and uric acid cause type 2 diabetes? Endocr. Rev. 2009, 30, 96–116. [Google Scholar] [CrossRef] [PubMed]
- Mayes, P.A. Intermediary metabolism of fructose. Am. J. Clin. Nutr. 1993, 58, 754S–765S. [Google Scholar] [PubMed]
- Douard, V.; Ferraris, R.P. Regulation of the fructose transporter GLUT5 in health and disease. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E227–E237. [Google Scholar] [CrossRef] [PubMed]
- Hajduch, E.; Darakhshan, F.; Hundal, H.S. Fructose uptake in rat adipocytes: GLUT5 expression and the effects of streptozotocin-induced diabetes. Diabetologia 1998, 41, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Heaney, A.P. Regulation of adipose differentiation by fructose and GluT5. Mol. Endocrinol. 2012, 26, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Froesch, E.R. Fructose metabolism in adipose tissue. Acta Med. Scand. Suppl. 1972, 542, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Varma, V.; Boros, L.G.; Nolen, G.T.; Chang, C.W.; Wabitsch, M.; Beger, R.D.; Kaput, J. Metabolic fate of fructose in human adipocytes: A targeted 13C tracer fate association study. Metabolomics 2015, 11, 529–544. [Google Scholar] [CrossRef] [PubMed]
- Varma, V.; Boros, L.G.; Nolen, G.T.; Chang, C.W.; Wabitsch, M.; Beger, R.D.; Kaput, J. Fructose Alters Intermediary Metabolism of Glucose in Human Adipocytes and Diverts Glucose to Serine Oxidation in the One-Carbon Cycle Energy Producing Pathway. Metabolites 2015, 5, 364–385. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, P.M.; Markert, E.K.; Gounder, M.; Lin, H.; Dvorzhinski, D.; Dolfi, S.C.; Chan, L.L.; Qiu, J.; DiPaola, R.S.; Hirshfield, K.M.; et al. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis. 2013, 4, e877. [Google Scholar] [CrossRef] [PubMed]
- Trommelen, J.; Fuchs, C.J.; Beelen, M.; Lenaerts, K.; Jeukendrup, A.E.; Cermak, N.M.; van Loon, L.J. Fructose and Sucrose Intake Increase Exogenous Carbohydrate Oxidation during Exercise. Nutrients 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.W.; Zachwieja, J.J.; Peronnet, F.; Passe, D.H.; Massicotte, D.; Lavoie, C.; Pascoe, D.D. Fuel selection and cycling endurance performance with ingestion of [13C]glucose: Evidence for a carbohydrate dose response. J. Appl. Physiol. 2010, 108, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Baur, D.A.; Schroer, A.B.; Luden, N.D.; Womack, C.J.; Smyth, S.A.; Saunders, M.J. Glucose-fructose enhances performance versus isocaloric, but not moderate, glucose. Med. Sci. Sports. Exerc. 2014, 46, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, D.S.; Swift, M.; Ros, M.; Green, J.G. Composite versus single transportable carbohydrate solution enhances race and laboratory cycling performance. Appl. Physiol. Nutr. Metab. 2012, 37, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Bally, L.; Kempf, P.; Zueger, T.; Speck, C.; Pasi, N.; Ciller, C.; Feller, K.; Loher, H.; Rosset, R.; Wilhelm, M.; et al. Metabolic Effects of Glucose-Fructose Co-Ingestion Compared to Glucose Alone during Exercise in Type 1 Diabetes. Nutrients 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Lecoultre, V.; Benoit, R.; Carrel, G.; Schutz, Y.; Millet, G.P.; Tappy, L.; Schneiter, P. Fructose and glucose co-ingestion during prolonged exercise increases lactate and glucose fluxes and oxidation compared with an equimolar intake of glucose. Am. J. Clin. Nutr. 2010, 92, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Sahebjami, H.; Scalettar, R. Effects of fructose infusion on lactate and uric acid metabolism. Lancet 1971, 1, 366–369. [Google Scholar] [CrossRef]
- Gregoire, F.M.; Smas, C.M.; Sul, H.S. Understanding adipocyte differentiation. Physiol. Rev. 1998, 78, 783–809. [Google Scholar] [PubMed]
- Lee, M.J.; Fried, S.K. The glucocorticoid receptor, not the mineralocorticoid receptor, plays the dominant role in adipogenesis and adipokine production in human adipocytes. Int. J. Obes. (Lond.) 2014, 38, 1228–1233. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. Molecular regulation of adipogenesis. Annu. Rev. Cell Dev. Biol. 2000, 16, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, L.; Smas, C.; Sul, H.S. Pref-1 interacts with fibronectin to inhibit adipocyte differentiation. Mol. Cell Biol. 2010, 30, 3480–3492. [Google Scholar] [CrossRef] [PubMed]
- De Sa, P.M.; Richard, A.J.; Hang, H.; Stephens, J.M. Transcriptional Regulation of Adipogenesis. Compr. Physiol. 2017, 7, 635–674. [Google Scholar]
- Poulos, S.P.; Dodson, M.V.; Culver, M.F.; Hausman, G.J. The increasingly complex regulation of adipocyte differentiation. Exp. Biol. Med. (Maywood) 2016, 241, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Nigro, D.; Chiazza, F.; Medana, C.; Bello, F.D.; Boccuzzi, G.; Collino, M.; Aragno, M. Fructose-derived advanced glycation end-products drive lipogenesis and skeletal muscle reprogramming via SREBP-1c dysregulation in mice. Free Radic. Biol. Med. 2016, 91, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Robubi, A.; Huber, K.R.; Krugluger, W. Extra fructose in the growth medium fuels lipogenesis of adipocytes. J. Obes. 2014, 2014, 647034. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, M.; Raji, L.; Prabhu, D.; Sathishkumar, C.; Prabu, P.; Mohan, V.; Balasubramanyam, M. High-fructose diet is as detrimental as high-fat diet in the induction of insulin resistance and diabetes mediated by hepatic/pancreatic endoplasmic reticulum (ER) stress. Mol. Cell Biochem. 2016, 423, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Petersen, K.F.; Shulman, G.I. Lipid-induced insulin resistance: Unravelling the mechanism. Lancet 2010, 375, 2267–2277. [Google Scholar] [CrossRef]
- Ye, J.; DeBose-Boyd, R.A. Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Petrie, J.L.; Patman, G.L.; Sinha, I.; Alexander, T.D.; Reeves, H.L.; Agius, L. The rate of production of uric acid by hepatocytes is a sensitive index of compromised cell ATP homeostasis. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1255–E1265. [Google Scholar] [CrossRef] [PubMed]
- Perheentupa, J.; Raivio, K. Fructose-induced hyperuricaemia. Lancet 1967, 2, 528–531. [Google Scholar] [CrossRef]
- Fiaschi, E.; Baggio, B.; Favaro, S.; Antonello, A.; Camerin, E.; Todesco, S.; Borsatti, A. Fructose-induced hyperuricemia in essential hypertension. Metabolism 1977, 26, 1219–1223. [Google Scholar] [CrossRef]
- Israel, K.D.; Michaelis, O.E.T.; Reiser, S.; Keeney, M. Serum uric acid, inorganic phosphorus, and glutamic-oxalacetic transaminase and blood pressure in carbohydrate-sensitive adults consuming three different levels of sucrose. Ann. Nutr. Metab. 1983, 27, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Reiser, S.; Powell, A.S.; Scholfield, D.J.; Panda, P.; Ellwood, K.C.; Canary, J.J. Blood lipids, lipoproteins, apoproteins, and uric acid in men fed diets containing fructose or high-amylose cornstarch. Am. J. Clin. Nutr. 1989, 49, 832–839. [Google Scholar] [PubMed]
- Odermatt, A. The Western-style diet: A major risk factor for impaired kidney function and chronic kidney disease. Am. J. Physiol. Renal. Physiol. 2011, 301, F919–F931. [Google Scholar] [CrossRef] [PubMed]
- Bos, M.J.; Koudstaal, P.J.; Hofman, A.; Witteman, J.C.; Breteler, M.M. Uric acid is a risk factor for myocardial infarction and stroke: The Rotterdam study. Stroke 2006, 37, 1503–1507. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Yang, F.; Yang, I.; Yin, Y.; Luo, J.J.; Wang, H.; Yang, X.F. Uric acid, hyperuricemia and vascular diseases. Front. Biosci. 2012, 17, 656–669. [Google Scholar] [CrossRef]
- Sanchez-Lozada, L.G.; Tapia, E.; Bautista-Garcia, P.; Soto, V.; Avila-Casado, C.; Vega-Campos, I.P.; Nakagawa, T.; Zhao, L.; Franco, M.; Johnson, R.J. Effects of febuxostat on metabolic and renal alterations in rats with fructose-induced metabolic syndrome. Am. J. Physiol. Renal. Physiol. 2008, 294, F710–F718. [Google Scholar] [CrossRef] [PubMed]
- Sautin, Y.Y.; Nakagawa, T.; Zharikov, S.; Johnson, R.J. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am. J. Physiol. Cell Physiol. 2007, 293, C584–C596. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.; Tzameli, I.; Pissios, P.; Rovira, I.; Gavrilova, O.; Ohtsubo, T.; Chen, Z.; Finkel, T.; Flier, J.S.; Friedman, J.M. Xanthine oxidoreductase is a regulator of adipogenesis and PPARgamma activity. Cell Metab. 2007, 5, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Stavric, B.; Johnson, W.J.; Clayman, S.; Gadd, R.E.; Chartrand, A. Effect of fructose administration on serum urate levels in the uricase inhibited rat. Experientia 1976, 32, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Kang, D.H.; Feng, L.; Nakagawa, T.; Kanellis, J.; Lan, H.; Mazzali, M.; Johnson, R.J. Uric acid, hominoid evolution, and the pathogenesis of salt-sensitivity. Hypertension 2002, 40, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Vasdev, S.; Gill, V.; Parai, S.; Longerich, L.; Gadag, V. Dietary vitamin E and C supplementation prevents fructose induced hypertension in rats. Mol. Cell Biochem. 2002, 241, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Kanellis, J.; Watanabe, S.; Li, J.H.; Kang, D.H.; Li, P.; Nakagawa, T.; Wamsley, A.; Sheikh-Hamad, D.; Lan, H.Y.; Feng, L.; et al. Uric acid stimulates monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-activated protein kinase and cyclooxygenase-2. Hypertension 2003, 41, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Gregor, M.F.; Hotamisligil, G.S. Thematic review series: Adipocyte Biology. Adipocyte stress: The endoplasmic reticulum and metabolic disease. J. Lipid. Res. 2007, 48, 1905–1914. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; MacDougald, O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006, 7, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Marek, G.; Pannu, V.; Shanmugham, P.; Pancione, B.; Mascia, D.; Crosson, S.; Ishimoto, T.; Sautin, Y.Y. Adiponectin resistance and proinflammatory changes in the visceral adipose tissue induced by fructose consumption via ketohexokinase-dependent pathway. Diabetes 2015, 64, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.V.; Schraw, T.D.; Kim, J.Y.; Khan, T.; Rajala, M.W.; Follenzi, A.; Scherer, P.E. Secretion of the adipocyte-specific secretory protein adiponectin critically depends on thiol-mediated protein retention. Mol. Cell Biol. 2007, 27, 3716–3731. [Google Scholar] [CrossRef] [PubMed]
- Diggle, C.P.; Shires, M.; Leitch, D.; Brooke, D.; Carr, I.M.; Markham, A.F.; Hayward, B.E.; Asipu, A.; Bonthron, D.T. Ketohexokinase: Expression and localization of the principal fructose-metabolizing enzyme. J. Histochem. Cytochem. 2009, 57, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Gao, J.; Ishigaki, Y.; Kondo, K.; Sawada, S.; Izumi, T.; Uno, K.; Kaneko, K.; Tsukita, S.; Takahashi, K.; et al. ER Stress Protein CHOP Mediates Insulin Resistance by Modulating Adipose Tissue Macrophage Polarity. Cell Rep. 2017, 18, 2045–2057. [Google Scholar] [CrossRef] [PubMed]
- Hoppmann, J.; Perwitz, N.; Meier, B.; Fasshauer, M.; Hadaschik, D.; Lehnert, H.; Klein, J. The balance between gluco—And mineralo-corticoid action critically determines inflammatory adipocyte responses. J. Endocrinol. 2010, 204, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.Y.; Daynes, R.A. Macrophages from 11beta-hydroxysteroid dehydrogenase type 1-deficient mice exhibit an increased sensitivity to lipopolysaccharide stimulation due to TGF-beta-mediated up-regulation of SHIP1 expression. J. Immunol. 2007, 179, 6325–6335. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, J.; Zhang, A.S.; Cheng, P.; Zhang, X.; Lv, S.; Wu, L.; Yu, J.; Di, W.J.; Zha, J.M.; et al. BVT.2733, a Selective 11 beta-Hydroxysteroid Dehydrogenase Type 1 Inhibitor, Attenuates Obesity and Inflammation in Diet-Induced Obese Mice. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Park, S.B.; Park, J.S.; Jung, W.H.; Kim, H.Y.; Kwak, H.J.; Ahn, J.H.; Choi, K.J.; Na, Y.J.; Choi, S.; Rhee, S.D.; et al. Anti-inflammatory effect of a selective 11beta-hydroxysteroid dehydrogenase type 1 inhibitor via the stimulation of heme oxygenase-1 in LPS-activated mice and J774.1 murine macrophages. J. Pharmacol. Sci. 2016, 131, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.J.J.; Thieringer, R.; Springer, M.S.; Wright, S.D.; Hermanowski-Vosatka, A.; Plump, A.; Balkovec, J.M.; Cheng, K.; Ding, G.J.; Kawka, D.W.; et al. 11 beta-HSD1 inhibition reduces atherosclerosis in mice by altering proinflammatory gene expression in the vasculature. Physiol. Genom. 2013, 45, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Chantong, B.; Kratschmar, D.V.; Nashev, L.G.; Balazs, Z.; Odermatt, A. Mineralocorticoid and glucocorticoid receptors differentially regulate NF-kappaB activity and pro-inflammatory cytokine production in murine BV-2 microglial cells. J. Neuroinflamm. 2012, 9, 260. [Google Scholar] [CrossRef] [PubMed]
- Hirata, A.; Maeda, N.; Nakatsuji, H.; Hiuge-Shimizu, A.; Okada, T.; Funahashi, T.; Shimomura, I. Contribution of glucocorticoid-mineralocorticoid receptor pathway on the obesity-related adipocyte dysfunction. Biochem. Biophys. Res. Commun. 2012, 419, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Farina, J.P.; Garcia, M.E.; Alzamendi, A.; Giovambattista, A.; Marra, C.A.; Spinedi, E.; Gagliardino, J.J. Antioxidant treatment prevents the development of fructose-induced abdominal adipose tissue dysfunction. Clin. Sci. 2013, 125, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Lavine, J.E.; Schwimmer, J.B.; van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. Nonalcoholic Steatohepatitis Clinical Research, Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: The TONIC randomized controlled trial. JAMA 2011, 305, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Bujalska, I.J.; Kumar, S.; Stewart, P.M. Does central obesity reflect ”Cushing’s disease of the omentum”? Lancet 1997, 349, 1210–1213. [Google Scholar] [CrossRef]
- Stewart, P.M. Tissue-specific Cushing’s syndrome, 11beta-hydroxysteroid dehydrogenases and the redefinition of corticosteroid hormone action. Eur. J. Endocrinol. 2003, 149, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Corbalan-Tutau, D.; Madrid, J.A.; Nicolas, F.; Garaulet, M. Daily profile in two circadian markers ”melatonin and cortisol” and associations with metabolic syndrome components. Physiol. Behav. 2014, 123, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, E.M.; Arregger, A.L.; Monardes, G.; Contreras, L.N. An accurate, non-invasive approach to diagnose Cushing’s syndrome in at-risk populations. Steroids 2013, 78, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, T.; Kulle, A.; Wolters, B.; Knop, C.; Lass, N.; Welzel, M.; Holterhus, P.M. Relationships between 24-hour urinary free cortisol concentrations and metabolic syndrome in obese children. J. Clin. Endocrinol. Metab. 2014, 99, 2391–2399. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, D.E.; Jones, G.C.; Smith, K.; Jamieson, P.M.; Andrew, R.; Kenyon, C.J.; Walker, B.R. Understanding the role of glucocorticoids in obesity: Tissue-specific alterations of corticosterone metabolism in obese Zucker rats. Endocrinology 2000, 141, 560–563. [Google Scholar] [CrossRef] [PubMed]
- Wake, D.J.; Strand, M.; Rask, E.; Westerbacka, J.; Livingstone, D.E.; Soderberg, S.; Andrew, R.; Yki-Jarvinen, H.; Olsson, T.; Walker, B.R. Intra-adipose sex steroid metabolism and body fat distribution in idiopathic human obesity. Clin. Endocrinol. (Oxf.) 2007, 66, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Rask, E.; Walker, B.R.; Soderberg, S.; Livingstone, D.E.; Eliasson, M.; Johnson, O.; Andrew, R.; Olsson, T. Tissue-specific changes in peripheral cortisol metabolism in obese women: Increased adipose 11beta-hydroxysteroid dehydrogenase type 1 activity. J. Clin. Endocrinol. Metab. 2002, 87, 3330–3336. [Google Scholar] [PubMed]
- Rask, E.; Olsson, T.; Soderberg, S.; Andrew, R.; Livingstone, D.E.; Johnson, O.; Walker, B.R. Tissue-specific dysregulation of cortisol metabolism in human obesity. J. Clin. Endocrinol. Metab. 2001, 86, 1418–1421. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.M.; Boulton, A.; Kumar, S.; Clark, P.M.; Shackleton, C.H. Cortisol metabolism in human obesity: Impaired cortisone→cortisol conversion in subjects with central adiposity. J. Clin. Endocrinol. Metab. 1999, 84, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- Westerbacka, J.; Yki-Jarvinen, H.; Vehkavaara, S.; Hakkinen, A.M.; Andrew, R.; Wake, D.J.; Seckl, J.R.; Walker, B.R. Body fat distribution and cortisol metabolism in healthy men: Enhanced 5beta-reductase and lower cortisol/cortisone metabolite ratios in men with fatty liver. J. Clin. Endocrinol. Metab. 2003, 88, 4924–4931. [Google Scholar] [CrossRef] [PubMed]
- Kotelevtsev, Y.; Holmes, M.C.; Burchell, A.; Houston, P.M.; Schmoll, D.; Jamieson, P.; Best, R.; Brown, R.; Edwards, C.R.; Seckl, J.R.; et al. 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc. Natl. Acad. Sci. USA 1997, 94, 14924–14929. [Google Scholar] [CrossRef] [PubMed]
- Marcolongo, P.; Senesi, S.; Giunti, R.; Csala, M.; Fulceri, R.; Banhegyi, G.; Benedetti, A. Expression of hexose-6-phosphate dehydrogenase in rat tissues. J. Steroid. Biochem. Mol. Biol. 2011, 126, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Marcolongo, P.; Senesi, S.; Gava, B.; Fulceri, R.; Sorrentino, V.; Margittai, E.; Lizak, B.; Csala, M.; Banhegyi, G.; Benedetti, A. Metyrapone prevents cortisone-induced preadipocyte differentiation by depleting luminal NADPH of the endoplasmic reticulum. Biochem. Pharmacol. 2008, 76, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Hauner, H.; Entenmann, G.; Wabitsch, M.; Gaillard, D.; Ailhaud, G.; Negrel, R.; Pfeiffer, E.F. Promoting effect of glucocorticoids on the differentiation of human adipocyte precursor cells cultured in a chemically defined medium. J. Clin. Investig. 1989, 84, 1663–1670. [Google Scholar] [CrossRef] [PubMed]
- Hauner, H.; Schmid, P.; Pfeiffer, E.F. Glucocorticoids and insulin promote the differentiation of human adipocyte precursor cells into fat cells. J. Clin. Endocrinol. Metab. 1987, 64, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.W.; Stewart, P.M. The functional consequences of 11beta-hydroxysteroid dehydrogenase expression in adipose tissue. Horm. Metab. Res. 2002, 34, 746–751. [Google Scholar] [CrossRef] [PubMed]
- Bujalska, I.J.; Kumar, S.; Hewison, M.; Stewart, P.M. Differentiation of adipose stromal cells: The roles of glucocorticoids and 11beta-hydroxysteroid dehydrogenase. Endocrinology 1999, 140, 3188–3196. [Google Scholar] [PubMed]
- Liu, Y.; Park, F.; Pietrusz, J.L.; Jia, G.; Singh, R.J.; Netzel, B.C.; Liang, M. Suppression of 11beta-hydroxysteroid dehydrogenase type 1 with RNA interference substantially attenuates 3T3-L1 adipogenesis. Physiol. Genom. 2008, 32, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Mandl, J.; Meszaros, T.; Banhegyi, G.; Hunyady, L.; Csala, M. Endoplasmic reticulum: Nutrient sensor in physiology and pathology. Trend Endocrinol. Metab. TEM 2009, 20, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Dzyakanchuk, A.A.; Balazs, Z.; Nashev, L.G.; Amrein, K.E.; Odermatt, A. 11beta-Hydroxysteroid dehydrogenase 1 reductase activity is dependent on a high ratio of NADPH/NADP(+) and is stimulated by extracellular glucose. Mol. Cell Endocrinol. 2009, 301, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Gumy, C.; Thurnbichler, C.; Aubry, E.M.; Balazs, Z.; Pfisterer, P.; Baumgartner, L.; Stuppner, H.; Odermatt, A.; Rollinger, J.M. Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 by plant extracts used as traditional antidiabetic medicines. Fitoterapia 2009, 80, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Froesch, E.R.; Ginsberg, J.L. Fructose metabolism of adipose tissue. I. Comparison of fructose and glucose metabolism in epididymal adipose tissue of normal rats. J. Biol. Chem. 1962, 237, 3317–3324. [Google Scholar] [PubMed]
- Apostolova, G.; Schweizer, R.A.; Balazs, Z.; Kostadinova, R.M.; Odermatt, A. Dehydroepiandrosterone inhibits the amplification of glucocorticoid action in adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E957–E964. [Google Scholar] [CrossRef] [PubMed]
- Balazs, Z.; Schweizer, R.A.; Frey, F.J.; Rohner-Jeanrenaud, F.; Odermatt, A. DHEA induces 11 -HSD2 by acting on CCAAT/enhancer-binding proteins. J. Am. Soc. Nephrol. 2008, 19, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Prince, P.D.; Santander, Y.; Gerez, E.M.; Hocht, C.; Polizio, A.H.; Mayer, M.A.; Taira, C.A.; Fraga, C.G.; Galleano, M.; Carranza, A. Fructose increases corticosterone production in association with NADPH metabolism alterations in rat epididymal white adipose tissue. J. Nutrit. Biochem. 2017. [Google Scholar] [CrossRef]
- Kovacevic, S.; Nestorov, J.; Matic, G.; Elakovic, I. Fructose and stress induce opposite effects on lipid metabolism in the visceral adipose tissue of adult female rats through glucocorticoid action. Europ. J. Nutr. 2016. [Google Scholar] [CrossRef]
- Bursac, B.N.; Djordjevic, A.D.; Vasiljevic, A.D.; Milutinovic, D.D.; Velickovic, N.A.; Nestorovic, N.M.; Matic, G.M. Fructose consumption enhances glucocorticoid action in rat visceral adipose tissue. J. Nutr. Biochem. 2013, 24, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Bursac, B.N.; Vasiljevic, A.D.; Nestorovic, N.M.; Velickovic, N.A.; Milutinovic, D.D.V.; Matic, G.M.; Djordjevic, A.D. High-fructose diet leads to visceral adiposity and hypothalamic leptin resistance in male rats—Do glucocorticoids play a role? J. Nutr. Biochem. 2014, 25, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Priyadarshini, E.; Anuradha, C.V. Glucocorticoid Antagonism Reduces Insulin Resistance and Associated Lipid Abnormalities in High-Fructose-Fed Mice. Can. J. Diabet. 2017, 41, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Larner, D.P.; Morgan, S.A.; Gathercole, L.L.; Doig, C.L.; Guest, P.; Weston, C.; Hazeldine, J.; Tomlinson, J.W.; Stewart, P.M.; Lavery, G.G. Male 11beta-HSD1 Knockout Mice Fed Trans-Fats and Fructose Are Not Protected From Metabolic Syndrome or Nonalcoholic Fatty Liver Disease. Endocrinology 2016, 157, 3493–3504. [Google Scholar] [CrossRef] [PubMed]
- Hellerstein, M.K. De novo lipogenesis in humans: Metabolic and regulatory aspects. Eur. J. Clin. Nutr. 1999, 53, S53–S65. [Google Scholar] [CrossRef] [PubMed]
- Baxter, J.D.; Forsham, P.H. Tissue effects of glucocorticoids. Am. J. Med. 1972, 53, 573–589. [Google Scholar] [CrossRef]
- Dolinsky, V.W.; Douglas, D.N.; Lehner, R.; Vance, D.E. Regulation of the enzymes of hepatic microsomal triacylglycerol lipolysis and re-esterification by the glucocorticoid dexamethasone. Biochem. J. 2004, 378, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.A.; McCabe, E.L.; Gathercole, L.L.; Hassan-Smith, Z.K.; Larner, D.P.; Bujalska, I.J.; Stewart, P.M.; Tomlinson, J.W.; Lavery, G.G. 11beta-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc. Natl. Acad Sci. USA 2014, 111, E2482–E2491. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Legeza, B.; Marcolongo, P.; Gamberucci, A.; Varga, V.; Bánhegyi, G.; Benedetti, A.; Odermatt, A. Fructose, Glucocorticoids and Adipose Tissue: Implications for the Metabolic Syndrome. Nutrients 2017, 9, 426. https://doi.org/10.3390/nu9050426
Legeza B, Marcolongo P, Gamberucci A, Varga V, Bánhegyi G, Benedetti A, Odermatt A. Fructose, Glucocorticoids and Adipose Tissue: Implications for the Metabolic Syndrome. Nutrients. 2017; 9(5):426. https://doi.org/10.3390/nu9050426
Chicago/Turabian StyleLegeza, Balázs, Paola Marcolongo, Alessandra Gamberucci, Viola Varga, Gábor Bánhegyi, Angiolo Benedetti, and Alex Odermatt. 2017. "Fructose, Glucocorticoids and Adipose Tissue: Implications for the Metabolic Syndrome" Nutrients 9, no. 5: 426. https://doi.org/10.3390/nu9050426
APA StyleLegeza, B., Marcolongo, P., Gamberucci, A., Varga, V., Bánhegyi, G., Benedetti, A., & Odermatt, A. (2017). Fructose, Glucocorticoids and Adipose Tissue: Implications for the Metabolic Syndrome. Nutrients, 9(5), 426. https://doi.org/10.3390/nu9050426