An In Vivo Magnetic Resonance Spectroscopy Study of the Effects of Caloric and Non-Caloric Sweeteners on Liver Lipid Metabolism in Rats

 , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Diets
2.2. MRS Experiments
2.3. Oral Glucose Tolerance Test
2.4. Plasma and Tissue Analyses
2.5. Determination of Glycogen Content in Liver
2.6. Glycolytic Enzyme Activities
2.7. Western Blot Analysis
2.8. Targeted Quantitative Mitochondrial Proteomics
2.9. Statistical Analysis
3. Results
3.1. Caloric Sweeteners Increase Adiposity without an Effect on Body Weight
3.2. Both Caloric and Non-Caloric Sweeteners Affect Whole-Body Glucose Homeostasis
3.3. Fructose Stimulates Hepatic De Novo Lipogenesis
3.4. Proteins Involved in Mitochondrial Oxidative Metabolism Are Differentially Affected by Glucose and Fructose Feeding
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A. Experimental Procedures
Western Blot Analysis

{kind=link}
{kind=link}
{kind=link}
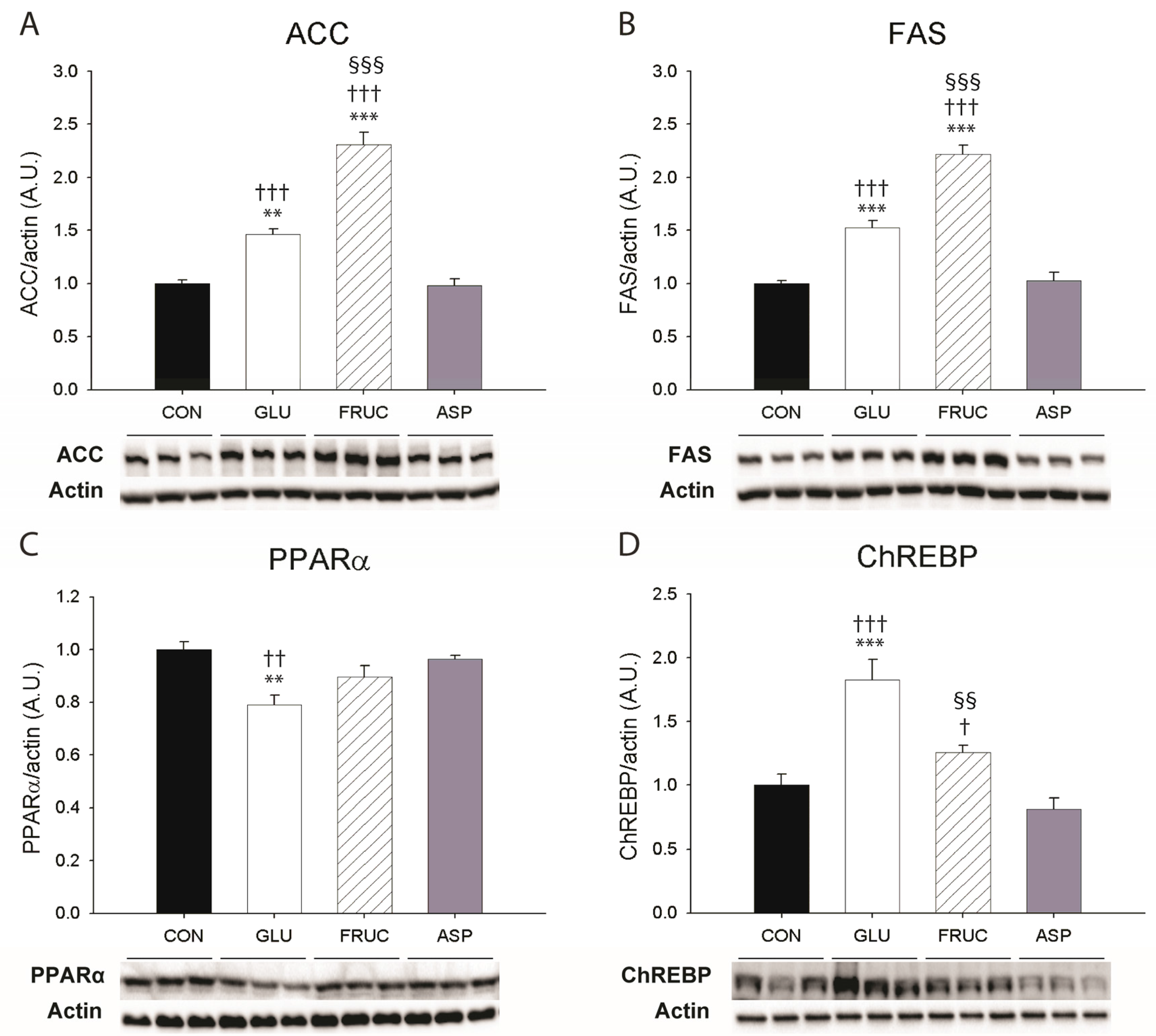{kind=link}
{kind=link}
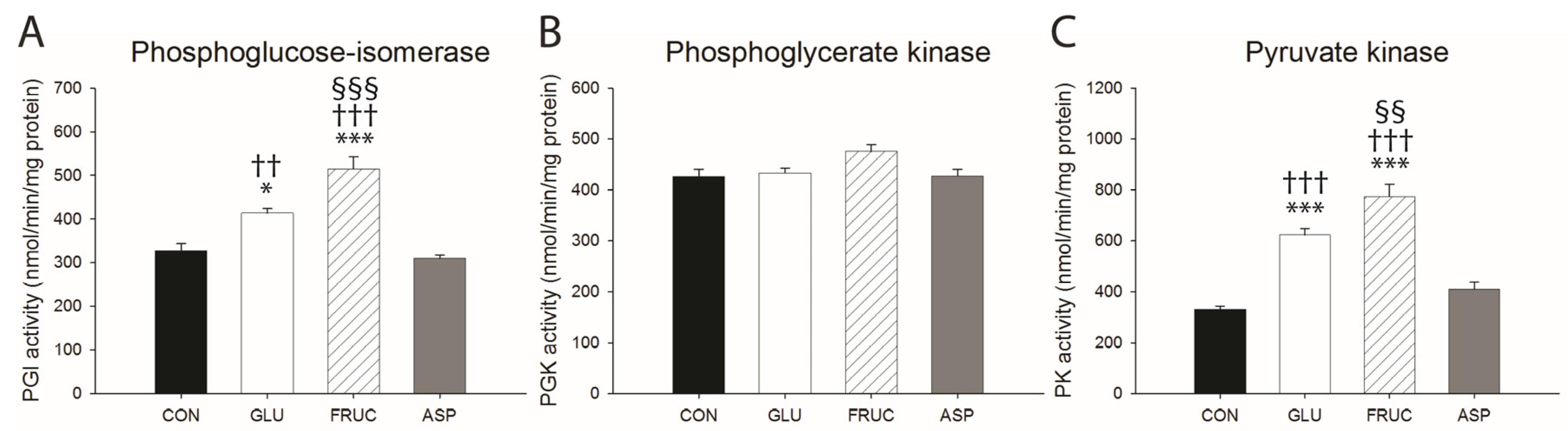{kind=link}
{kind=link}
| Gene Name | Protein Name | CON | GLU | FRUC | ASP | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fatty acid β-oxidation | |||||||||||||
| Acaa2 | Acetyl-CoA acyltransferase 2 | 34.2 | ± | 0.7 | 32.5 | ± | 1.2 † | 36.5 | ± | 2.3 | 39.0 | ± | 1.7 |
| Acadl | Acyl-CoA dehydrogenase, long-chain | 38.7 | ± | 1.3 | 34.9 | ± | 1.6 | 39.0 | ± | 2.1 | 41.0 | ± | 1.9 |
| Acads | Acyl-CoA dehydrogenase, short-chain | 11.4 | ± | 0.5 | 10.5 | ± | 0.2 | 10.9 | ± | 0.3 | 10.1 | ± | 0.4 |
| Acadvl | Acyl-CoA dehydrogenase, very long-chain | 12.5 | ± | 0.6 | 11.5 | ± | 0.2 | 10.1 | ± | 0.6 * | 11.0 | ± | 0.5 |
| Cpt1a | Carnitine O-palmitoyltransferase 1A | 8.0 | ± | 0.2 | 5.7 | ± | 0.4 ** | 5.8 | ± | 0.4 ** | 7.0 | ± | 0.4 |
| Cpt1b | Carnitine O-palmitoyltransferase 1B | 1.29 | ± | 0.07 | 1.96 | ± | 0.19 | 1.46 | ± | 0.12 | 1.65 | ± | 0.38 |
| Cpt2 | Carnitine palmitoyltransferase 2 | 6.2 | ± | 0.2 | 6.8 | ± | 0.3 | 6.5 | ± | 0.6 | 5.4 | ± | 0.3 |
| Decr1 | 2,4-dienoyl CoA reductase 1 | 7.8 | ± | 0.22 | 7.4 | ± | 0.3 †† | 8.3 | ± | 0.7 † | 10.0 | ± | 0.2 ** |
| Echs1 | Enoyl CoA hydratase, short chain, 1 | 20.0 | ± | 0.8 | 16.0 | ± | 0.7 *,††† | 18.7 | ± | 1.5 †† | 24.1 | ± | 0.9 * |
| Eci1 | Enoyl-CoA delta isomerase | 11.9 | ± | 0.7 | 11.8 | ± | 0.7 | 12.0 | ± | 1.0 | 13.1 | ± | 0.6 |
| Etfa | Electron-transfer-flavoprotein, alpha polypeptide | 18.9 | ± | 0.8 | 15.4 | ± | 1.0 †† | 19.5 | ± | 1.8 | 23.3 | ± | 1.3 |
| Etfb | Electron-transfer-flavoprotein, beta polypeptide | 25.1 | ± | 0.8 | 20.1 | ± | 1.0 **,†† | 23.9 | ± | 1.0 | 25.0 | ± | 1.0 |
| Etfdh | Electron-transferring-flavoprotein dehydrogenase | 15.5 | ± | 0.4 | 13.3 | ± | 0.6 ** | 14.1 | ± | 0.4 | 14.6 | ± | 0.3 |
| Hadh | Hydroxyacyl-CoA dehydrogenase | 34.4 | ± | 1.1 | 30.9 | ± | 1.2 | 32.7 | ± | 1.2 | 34.0 | ± | 1.2 |
| Hadha | Hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase (trifunctional protein), alpha subunit | 16.1 | ± | 0.3 | 18.3 | ± | 0.4 *,††† | 17.1 | ± | 0.6 ††† | 13.5 | ± | 0.6 ** |
| Hadhb | Hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase (trifunctional protein), beta subunit | 13.6 | ± | 0.4 | 16.5 | ± | 0.3 **,††† | 15.3 | ± | 0.5 ††† | 12.0 | ± | 0.4 |
| Slc25a20 | Solute carrier family 25, member 20 | 4.1 | ± | 0.2 | 3.1 | ± | 0.3 ††† | 4.4 | ± | 0.3 § | 5.4 | ± | 0.4 * |
| TCA cycle | |||||||||||||
| Aco2 | Aconitase 2 | 11.1 | ± | 0.4 | 9.9 | ± | 0.2 * | 11.4 | ± | 0.1 §§,† | 10.1 | ± | 0.3 |
| Cs | Citrate synthase | 5.40 | ± | 0.13 | 4.66 | ± | 0.14 †† | 5.59 | ± | 0.39 § | 6.04 | ± | 0.09 |
| Dlat | Dihydrolipoamide S-acetyltransferase | 7.1 | ± | 0.2 | 8.7 | ± | 0.5 *,†† | 10.3 | ± | 0.3 ***,§§,††† | 6.8 | ± | 0.2 |
| Dld | Dihydrolipoamide dehydrogenase | 23.3 | ± | 0.7 | 21.2 | ± | 1.1 | 23.3 | ± | 1.1 | 20.7 | ± | 0.4 |
| Dlst | Dihydrolipoamide S-succinyltransferase (E2 component of 2-oxo-glutarate complex) | 13.4 | ± | 0.8 | 11.7 | ± | 0.6 | 13.2 | ± | 0.2 | 13.3 | ± | 0.4 |
| Fh1 | Fumarate hydratase 1 | 14.1 | ± | 0.7 | 12.1 | ± | 0.6 | 15.9 | ± | 1.5 | 16.0 | ± | 1.0 |
| Idh2 | Isocitrate dehydrogenase 2 (NADP+) | 10.0 | ± | 0.4 | 9.2 | ± | 0.6 | 9.2 | ± | 0.2 | 9.0 | ± | 0.3 |
| Idh3a | Isocitrate dehydrogenase [NAD] subunit α | 3.3 | ± | 0.4 | 3.1 | ± | 0.5 | 2.7 | ± | 0.2 | 3.2 | ± | 0.2 |
| Mdh2 | Malate dehydrogenase 2 | 31.6 | ± | 1.0 | 27.2 | ± | 0.9 † | 31.2 | ± | 2.0 | 33.8 | ± | 1.6 |
| Ogdh | Oxoglutarate (alpha-ketoglutarate) dehydrogenase | 6.1 | ± | 0.1 | 5.3 | ± | 0.2 * | 5.7 | ± | 0.2 | 5.3 | ± | 0.2 * |
| Pdha1 | Pyruvate dehydrogenase E1 component subunit alpha | 6.1 | ± | 0.2 | 7.8 | ± | 0.4 * | 9.4 | ± | 0.3 ***,§,†† | 7.3 | ± | 0.5 |
| Pdk1 | Pyruvate dehydrogenase kinase, isozyme 1 | 1.06 | ± | 0.01 | 1.06 | ± | 0.03 †† | 1.17 | ± | 0.02 **,†† | 1.05 | ± | 0.02 |
| Slc25a1 | Solute carrier family 25, member 1 | 11.8 | ± | 0.4 | 10.2 | ± | 0.4 * | 11.7 | ± | 0.3 | 11.6 | ± | 0.5 |
| Slc25a10 | Solute carrier family 25, member 10 | 9.6 | ± | 0.5 | 6.8 | ± | 0.5 **,†† | 8.0 | ± | 0.4 | 9.5 | ± | 0.6 |
| Slc25a11 | Solute carrier family 25, member 11 | 5.5 | ± | 0.4 | 4.6 | ± | 0.2 | 4.7 | ± | 0.4 | 5.1 | ± | 0.3 |
| Slc25a22 | Solute carrier family 25, member 22 | 3.99 | ± | 0.11 | 3.31 | ± | 0.09 **,† | 3.22 | ± | 0.11 **,†† | 3.85 | ± | 0.15 |
| Sucla2 | Succinyl-CoA ligase [ADP-forming] subunit beta | 8.2 | ± | 0.3 | 6.5 | ± | 0.2 ***,†† | 6.9 | ± | 0.2 ** | 7.8 | ± | 0.3 |
| Suclg1 | Succinate-CoA ligase, alpha subunit | 18.9 | ± | 0.6 | 15.2 | ± | 0.5 **,† | 16.3 | ± | 0.7 * | 18.5 | ± | 0.7 |
| Suclg2 | Succinate-CoA ligase, beta subunit | 11.6 | ± | 0.3 | 10.3 | ± | 0.3 †† | 10.9 | ± | 0.2 † | 12.7 | ± | 0.6 |
| Oxidative phosphorylation | |||||||||||||
| Ndufs1 | NADH dehydrogenase (ubiquinone) Fe-S protein 1 | 5.4 | ± | 0.3 | 4.5 | ± | 0.2 * | 5.8 | ± | 0.2 §§ | 5.1 | ± | 0.2 |
| Sdha | Succinate dehydrogenase complex, subunit A, flavoprotein | 10.0 | ± | 0.3 | 8.9 | ± | 0.3 * | 9.7 | ± | 0.2 | 9.9 | ± | 0.3 |
| Sdhb | Succinate dehydrogenase complex, subunit B, iron sulfur (Ip) | 6.8 | ± | 0.2 | 5.1 | ± | 0.1 *,††† | 6.9 | ± | 0.5 §§ | 7.4 | ± | 0.3 |
| Uqcrc2 | Ubiquinol-cytochrome c reductase core protein II | 8.5 | ± | 0.3 | 7.3 | ± | 0.3 ††† | 9.1 | ± | 0.5 § | 9.9 | ± | 0.4 |
| Cox5a | Cytochrome c oxidase subunit Va | 5.31 | ± | 0.24 | 4.86 | ± | 0.23 †† | 5.70 | ± | 0.08 § | 5.79 | ± | 0.11 |
| Cycs | Cytochrome c, somatic | 3.26 | ± | 0.25 | 2.94 | ± | 0.14 | 2.88 | ± | 0.09 | 3.13 | ± | 0.13 |
| Atp5b | ATP synthase, H+ transporting, mitochondrial F1 complex, beta polypeptide | 63.2 | ± | 2.1 | 58.4 | ± | 2.5 † | 71.5 | ± | 5.2 § | 73.0 | ± | 2.1 |
| Slc25a3 | Solute carrier family 25 (mitochondrial carrier; phosphate carrier), member 3 | 22.4 | ± | 0.7 | 17.2 | ± | 0.5 *** | 19.3 | ± | 0.9 * | 19.0 | ± | 0.8 * |
| Slc25a4 | Solute carrier family 25 (mitochondrial carrier; phosphate carrier), member 4 | 0.97 | ± | 0.04 | 0.87 | ± | 0.05 | 0.86 | ± | 0.02 | 0.83 | ± | 0.03 |
| Slc25a5 | Solute carrier family 25 (mitochondrial carrier; phosphate carrier), member 5 | 49.4 | ± | 1.7 | 39.8 | ± | 1.3 ** | 43.6 | ± | 1.9 | 42.7 | ± | 1.1 * |
| Ucp2 | Uncoupling protein 2 | 0.80 | ± | 0.04 | 0.84 | ± | 0.06 | 0.70 | ± | 0.04 | 0.71 | ± | 0.06 |
| Ucp3 | Uncoupling protein 3 | 0.92 | ± | 0.04 | 1.08 | ± | 0.12 † | 0.81 | ± | 0.04 | 0.78 | ± | 0.05 |
References
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the global burden of disease study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Berlanga, A.; Guiu-Jurado, E.; Porras, J.A.; Auguet, T. Molecular pathways in non-alcoholic fatty liver disease. Clin. Exp. Gastroenterol. 2014, 7, 221–239. [Google Scholar] [PubMed]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A.; Nielsen, S.J.; Popkin, B.M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004, 79, 537–543. [Google Scholar] [PubMed]
- Bray, G.A. Soft drink consumption and obesity: It is all about fructose. Curr. Opin. Lipidol. 2010, 21, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A. Carbohydrate intake and nonalcoholic fatty liver disease. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Le, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T. Fructose induced lipogenesis: From sugar to fat to insulin resistance. Trends Endocrinol. Metab. 2011, 22, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Schwarz, J.M.; Havel, P.J. Adverse metabolic effects of dietary fructose: Results from the recent epidemiological, clinical, and mechanistic studies. Curr. Opin. Lipidol. 2013, 24, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.Y.; Miyashita, M.; Cho, B.H.; Nakamura, M.T. Replacing dietary glucose with fructose increases ChREBP activity and SREBP-1 protein in rat liver nucleus. Biochem. Biophys. Res. Commun. 2009, 390, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Janevski, M.; Ratnayake, S.; Siljanovski, S.; McGlynn, M.A.; Cameron-Smith, D.; Lewandowski, P. Fructose containing sugars modulate mRNA of lipogenic genes ACC and FAS and protein levels of transcription factors ChREBP and SREBP1c with no effect on body weight or liver fat. Food Funct. 2012, 3, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, A.; Roglans, N.; Baena, M.; Sanchez, R.M.; Merlos, M.; Alegret, M.; Laguna, J.C. Liquid fructose downregulates Sirt1 expression and activity and impairs the oxidation of fatty acids in rat and human liver cells. Biochim. Biophys. Acta 2014, 1841, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, M.; Lin, X.; Li, Y.; Liu, C.; Yang, Y.; Yamahara, J.; Wang, J. Modulation of hepatic sterol regulatory element-binding protein-1c-mediated gene expression contributes to Salacia oblonga root-elicited improvement of fructose-induced fatty liver in rats. J. Ethnopharmacol. 2013, 150, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Rizkalla, S.W. Health implications of fructose consumption: A review of recent data. Nutr. Metab. Lond. 2010, 7, 82. [Google Scholar] [CrossRef] [PubMed]
- Roglans, N.; Vila, L.; Farre, M.; Alegret, M.; Sanchez, R.M.; Vazquez-Carrera, M.; Laguna, J.C. Impairment of hepatic Stat-3 activation and reduction of PPARα activity in fructose-fed rats. Hepatology 2007, 45, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Parks, E.J.; Krauss, R.M.; Christiansen, M.P.; Neese, R.A.; Hellerstein, M.K. Effects of a low-fat, high-carbohydrate diet on VLDL-triglyceride assembly, production, and clearance. J. Clin. Investig. 1999, 104, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Haidari, M.; Leung, N.; Mahbub, F.; Uffelman, K.D.; Kohen-Avramoglu, R.; Lewis, G.F.; Adeli, K. Fasting and postprandial overproduction of intestinally derived lipoproteins in an animal model of insulin resistance. Evidence that chronic fructose feeding in the hamster is accompanied by enhanced intestinal de novo lipogenesis and Apob48-containing lipoprotein overproduction. J. Biol. Chem. 2002, 277, 31646–31655. [Google Scholar] [PubMed]
- Lewis, G.F.; Uffelman, K.; Naples, M.; Szeto, L.; Haidari, M.; Adeli, K. Intestinal lipoprotein overproduction, a newly recognized component of insulin resistance, is ameliorated by the insulin sensitizer rosiglitazone: Studies in the fructose-fed Syrian golden hamster. Endocrinology 2005, 146, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Raychaudhuri, U.; Chakraborty, R. Artificial sweeteners—A review. J. Food Sci. Technol. 2014, 51, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Mitsutomi, K.; Masaki, T.; Shimasaki, T.; Gotoh, K.; Chiba, S.; Kakuma, T.; Shibata, H. Effects of a nonnutritive sweetener on body adiposity and energy metabolism in mice with diet-induced obesity. Metabolism 2014, 63, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Anton, S.D.; Martin, C.K.; Han, H.; Coulon, S.; Cefalu, W.T.; Geiselman, P.; Williamson, D.A. Effects of stevia, aspartame, and sucrose on food intake, satiety, and postprandial glucose and insulin levels. Appetite 2010, 55, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Raben, A.; Vasilaras, T.H.; Moller, A.C.; Astrup, A. Sucrose compared with artificial sweeteners: Different effects on ad libitum food intake and body weight after 10 weeks of supplementation in overweight subjects. Am. J. Clin. Nutr. 2002, 76, 721–729. [Google Scholar] [PubMed]
- Suez, J.; Korem, T.; Zeevi, D.; Zilberman-Schapira, G.; Thaiss, C.A.; Maza, O.; Israeli, D.; Zmora, N.; Gilad, S.; Weinberger, A.; et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 2014, 514, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Bellisle, F.; Drewnowski, A. Intense sweeteners, energy intake and the control of body weight. Eur. J. Clin. Nutr. 2007, 61, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Davidson, T.L.; Martin, A.A.; Clark, K.; Swithers, S.E. Intake of high-intensity sweeteners alters the ability of sweet taste to signal caloric consequences: Implications for the learned control of energy and body weight regulation. Q. J. Exp. Psychol. Hove 2011, 64, 1430–1441. [Google Scholar] [CrossRef] [PubMed]
- Swithers, S.E. Artificial sweeteners produce the counterintuitive effect of inducing metabolic derangements. Trends Endocrinol. Metab. 2013, 24, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Gul, S.S.; Hamilton, A.R.L.; Munoz, A.R.; Phupitakphol, T.; Liu, W.; Hyoju, S.K.; Economopoulos, K.P.; Morrison, S.; Hu, D.; Zhang, W.F.; et al. Inhibition of the gut enzyme intestinal alkaline phosphatase may explain how aspartame promotes glucose intolerance and obesity in mice. Appl. Physiol. Nutr. Metab. 2017, 42, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Palmnas, M.S.; Cowan, T.E.; Bomhof, M.R.; Su, J.; Reimer, R.A.; Vogel, H.J.; Hittel, D.S.; Shearer, J. Low-dose aspartame consumption differentially affects gut microbiota-host metabolic interactions in the diet-induced obese rat. PLoS ONE 2014, 9, e109841. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, R.A.; Geraedts, T.R.; van Loon, L.J.; Nicolay, K.; Prompers, J.J. Multitissue assessment of in vivo postprandial intracellular lipid partitioning in rats using localized 1H-[13C] magnetic resonance spectroscopy. Magn. Reson. Med. 2012, 68, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, R.A.; van Loon, L.J.; Nicolay, K.; Prompers, J.J. In vivo postprandial lipid partitioning in liver and skeletal muscle in prediabetic and diabetic rats. Diabetologia 2013, 56, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Demoz, A.; Garras, A.; Asiedu, D.K.; Netteland, B.; Berge, R.K. Rapid method for the separation and detection of tissue short-chain coenzyme a esters by reversed-phase high-performance liquid chromatography. J. Chromatogr. B Biomed. Appl. 1995, 667, 148–152. [Google Scholar] [CrossRef]
- Van Hoek, P.; Flikweert, M.T.; van der Aart, Q.J.; Steensma, H.Y.; van Dijken, J.P.; Pronk, J.T. Effects of pyruvate decarboxylase overproduction on flux distribution at the pyruvate branch point in saccharomyces cerevisiae. Appl. Environ. Microbiol. 1998, 64, 2133–2140. [Google Scholar] [PubMed]
- Van Hoek, P.; van Dijken, J.P.; Pronk, J.T. Regulation of fermentative capacity and levels of glycolytic enzymes in chemostat cultures of saccharomyces cerevisiae. Enzym. Microb. Technol. 2000, 26, 724–736. [Google Scholar] [CrossRef]
- Van Hoek, P.; Van Dijken, J.P.; Pronk, J.T. Effect of specific growth rate on fermentative capacity of baker’s yeast. Appl. Environ. Microbiol. 1998, 64, 4226–4233. [Google Scholar] [PubMed]
- Wolters, J.C.; Ciapaite, J.; van Eunen, K.; Niezen-Koning, K.E.; Matton, A.; Porte, R.J.; Horvatovich, P.; Bakker, B.M.; Bischoff, R.; Permentier, H.P. Translational targeted proteomics profiling of mitochondrial energy metabolic pathways in mouse and human samples. J. Proteome Res. 2016, 15, 3204–3213. [Google Scholar] [CrossRef] [PubMed]
- Ventura, E.E.; Davis, J.N.; Goran, M.I. Sugar content of popular sweetened beverages based on objective laboratory analysis: Focus on fructose content. Obesity 2011, 19, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Park, O.J.; Cesar, D.; Faix, D.; Wu, K.; Shackleton, C.H.; Hellerstein, M.K. Mechanisms of fructose-induced hypertriglyceridaemia in the rat. Activation of hepatic pyruvate dehydrogenase through inhibition of pyruvate dehydrogenase kinase. Biochem. J. 1992, 282, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Peredo, H.A.; Lee, H.; Donoso, A.S.; Andrade, V.; Sanchez Eluchans, N.; Puyo, A.M. A high-fat plus fructose diet produces a vascular prostanoid alterations in the rat. Auton. Autacoid Pharmacol. 2015, 34, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.C.; Francini, F.; Gagliardino, J.J.; Massa, M.L. Lipoic acid prevents fructose-induced changes in liver carbohydrate metabolism: Role of oxidative stress. Biochim. Biophys. Acta 2014, 1840, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Sheludiakova, A.; Rooney, K.; Boakes, R.A. Metabolic and behavioural effects of sucrose and fructose/glucose drinks in the rat. Eur. J. Nutr. 2012, 51, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Thibault, L.; Woods, S.C.; Westerterp-Plantenga, M.S. The utility of animal models of human energy homeostasis. Br. J. Nutr. 2004, 92, S41–S45. [Google Scholar] [CrossRef] [PubMed]
- Kuzma, J.N.; Cromer, G.; Hagman, D.K.; Breymeyer, K.L.; Roth, C.L.; Foster-Schubert, K.E.; Holte, S.E.; Callahan, H.S.; Weigle, D.S.; Kratz, M. No difference in ad libitum energy intake in healthy men and women consuming beverages sweetened with fructose, glucose, or high-fructose corn syrup: A randomized trial. Am. J. Clin. Nutr. 2015, 102, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.Y.; Wallig, M.A.; Chung, B.H.; Nara, T.Y.; Cho, B.H.; Nakamura, M.T. Dietary fructose induces a wide range of genes with distinct shift in carbohydrate and lipid metabolism in fed and fasted rat liver. Biochim. Biophys. Acta 2008, 1782, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Nunes, P.M.; Wright, A.J.; Veltien, A.; van Asten, J.J.; Tack, C.J.; Jones, J.G.; Heerschap, A. Dietary lipids do not contribute to the higher hepatic triglyceride levels of fructose-compared to glucose-fed mice. FASEB J. 2014, 28, 1988–1997. [Google Scholar] [CrossRef] [PubMed]
- Lecoultre, V.; Egli, L.; Carrel, G.; Theytaz, F.; Kreis, R.; Schneiter, P.; Boss, A.; Zwygart, K.; Le, K.A.; Bortolotti, M.; et al. Effects of fructose and glucose overfeeding on hepatic insulin sensitivity and intrahepatic lipids in healthy humans. Obesity 2013, 21, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Vila, L.; Roglans, N.; Perna, V.; Sanchez, R.M.; Vazquez-Carrera, M.; Alegret, M.; Laguna, J.C. Liver AMP/ATP ratio and fructokinase expression are related to gender differences in AMPK activity and glucose intolerance in rats ingesting liquid fructose. J. Nutr. Biochem. 2011, 22, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Hagopian, K.; Chen, Y.; Simmons Domer, K.; Soo Hoo, R.; Bentley, T.; McDonald, R.B.; Ramsey, J.J. Caloric restriction influences hydrogen peroxide generation in mitochondrial sub-populations from mouse liver. J. Bioenerg. Biomembr. 2011, 43, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Dukhande, V.V.; Sharma, G.C.; Lai, J.C.; Farahani, R. Chronic hypoxia-induced alterations of key enzymes of glucose oxidative metabolism in developing mouse liver are mTOR dependent. Mol. Cell. Biochem. 2011, 357, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.C.; Lin, Y.L.; Kuo, C.F. Effect of high-fat diet on hepatic proteomics of hamsters. J. Agric. Food Chem. 2015, 63, 1869–1881. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.; Bickerton, A.S.; Fielding, B.A.; Blaak, E.E.; Wagenmakers, A.J.; Chong, M.F.; Gilbert, M.; Karpe, F.; Frayn, K.N. Reduced oxidation of dietary fat after a short term high-carbohydrate diet. Am. J. Clin. Nutr. 2008, 87, 824–831. [Google Scholar] [PubMed]
- Chong, M.F.; Fielding, B.A.; Frayn, K.N. Mechanisms for the acute effect of fructose on postprandial lipemia. Am. J. Clin. Nutr. 2007, 85, 1511–1520. [Google Scholar] [PubMed]







| CON | GLU | FRUC | ASP | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Start body weight (g) | 354 | ± | 6 | 347 | ± | 3 | 351 | ± | 3 | 348 | ± | 3 |
| End body weight (g) | 445 | ± | 5 | 444 | ± | 8 | 456 | ± | 5 | 437 | ± | 6 |
| Body weight gain (g) | 92 | ± | 4 | 97 | ± | 6 | 105 | ± | 4 | 89 | ± | 4 |
| Food intake (kJ/week) | 2300 | ± | 25 | 1170 | ± | 24 ***,††† | 1447 | ± | 36 ***,†††,§§§ | 2232 | ± | 35 |
| Food intake (g/week) | 180 | ± | 2 | 91 | ± | 2 ***,††† | 113 | ± | 3 ***,†††,§§§ | 174 | ± | 3 |
| Drink intake (kJ/week) | NA | 1757.9 | ± | 48.7 ††† | 1274.4 | ± | 44.9 †††,§§§ | 13.4 | ± | 0.5 | ||
| Drink intake (mL/week) | 177 | ± | 4 | 808 | ± | 22 ***,††† | 586 | ± | 21 ***,†††,§§§ | 201 | ± | 7 |
| Total energy intake (kJ/week) | 2300 | ± | 25 | 2927 | ± | 35 ***,††† | 2721 | ± | 20 ***,†††,§§§ | 2246 | ± | 36 |
| Amount sweetener (g/kg BW/day) | NA | 36.87 | ± | 1.25 ††† | 26.16 | ± | 1.00 †††,§§§ | 0.28 | ± | 0.01 | ||
| Epididymal fat (g) (n = 36) | 5.8 | ± | 0.2 | 7.5 | ± | 0.4 **,†† | 7.1 | ± | 0.3 *,† | 5.6 | ± | 0.3 |
| Perirenal fat (g) (n = 36) | 5.8 | ± | 0.4 | 9.5 | ± | 0.6 ***,††† | 9.0 | ± | 0.5 ***,††† | 5.8 | ± | 0.4 |
| Plasma TG (mM) | 1.05 | ± | 0.10 | 1.06 | ± | 0.10 | 1.23 | ± | 0.04 † | 0.87 | ± | 0.04 |
| Plasma ALT (U/L) | 24.94 | ± | 2.35 | 21.71 | ± | 3.11 | 51.21 | ± | 8.00 **,§§ | 35.44 | ± | 4.32 |
| Experimental group 1 (n = 9 per diet group) | ||||||||||||
| Liver weight (g) | 10.63 | ± | 0.14 | 9.97 | ± | 0.16 | 11.39 | ± | 0.22 †,§§ | 10.17 | ± | 0.25 |
| Liver glycogen (mg/g ww) | 87 | ± | 10 | 78 | ± | 7 | 75 | ± | 4 | 67 | ± | 6 |
| Experimental group 2 (n = 6 per diet group) | ||||||||||||
| Liver weight (g) | 12.92 | ± | 0.16 ### | 13.08 | ± | 0.64 ### | 16.25 | ± | 0.59 ***,†††,§§§,### | 13.23 | ± | 0.21 ### |
| Liver glycogen (mg/g ww) | 96 | ± | 3 | 118 | ± | 8 ### | 117 | ± | 8 ### | 115 | ± | 8 ### |
| Liver malonyl-CoA (nmol/g ww) | 58.37 | ± | 0.97 | 62.44 | ± | 2.70 | 68.05 | ± | 2.32 *,†† | 56.31 | ± | 1.82 |
| CON | GLU | FRUC | ASP | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fasting glucose (mM) | 4.87 | ± | 0.21 | 4.44 | ± | 0.27 | 4.35 | ± | 0.10 | 5.22 | ± | 0.40 |
| AUCg (mM·h) | 13.67 | ± | 0.39 | 15.07 | ± | 0.55 | 14.89 | ± | 0.43 | 13.87 | ± | 0.46 |
| Fasting insulin (pM) | 267 | ± | 64 | 605 | ± | 97 | 500 | ± | 81 | 573 | ± | 61 * |
| AUCi (pM·h) | 454 | ± | 64 | 576 | ± | 75 | 614 | ± | 54 | 562 | ± | 52 |
| AUGg·AUCi (mM·h·pM·h) | 5462 | ± | 672 | 8640 | ± | 1151 | 9274 | ± | 1031 * | 7824 | ± | 770 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janssens, S.; Ciapaite, J.; Wolters, J.C.; Van Riel, N.A.; Nicolay, K.; Prompers, J.J. An In Vivo Magnetic Resonance Spectroscopy Study of the Effects of Caloric and Non-Caloric Sweeteners on Liver Lipid Metabolism in Rats. Nutrients 2017, 9, 476. https://doi.org/10.3390/nu9050476
Janssens S, Ciapaite J, Wolters JC, Van Riel NA, Nicolay K, Prompers JJ. An In Vivo Magnetic Resonance Spectroscopy Study of the Effects of Caloric and Non-Caloric Sweeteners on Liver Lipid Metabolism in Rats. Nutrients. 2017; 9(5):476. https://doi.org/10.3390/nu9050476
Chicago/Turabian StyleJanssens, Sharon, Jolita Ciapaite, Justina C. Wolters, Natal A. Van Riel, Klaas Nicolay, and Jeanine J. Prompers. 2017. "An In Vivo Magnetic Resonance Spectroscopy Study of the Effects of Caloric and Non-Caloric Sweeteners on Liver Lipid Metabolism in Rats" Nutrients 9, no. 5: 476. https://doi.org/10.3390/nu9050476
APA StyleJanssens, S., Ciapaite, J., Wolters, J. C., Van Riel, N. A., Nicolay, K., & Prompers, J. J. (2017). An In Vivo Magnetic Resonance Spectroscopy Study of the Effects of Caloric and Non-Caloric Sweeteners on Liver Lipid Metabolism in Rats. Nutrients, 9(5), 476. https://doi.org/10.3390/nu9050476