Toxic AGE (TAGE) Theory for the Pathophysiology of the Onset/Progression of NAFLD and ALD

Abstract
:1. Introduction
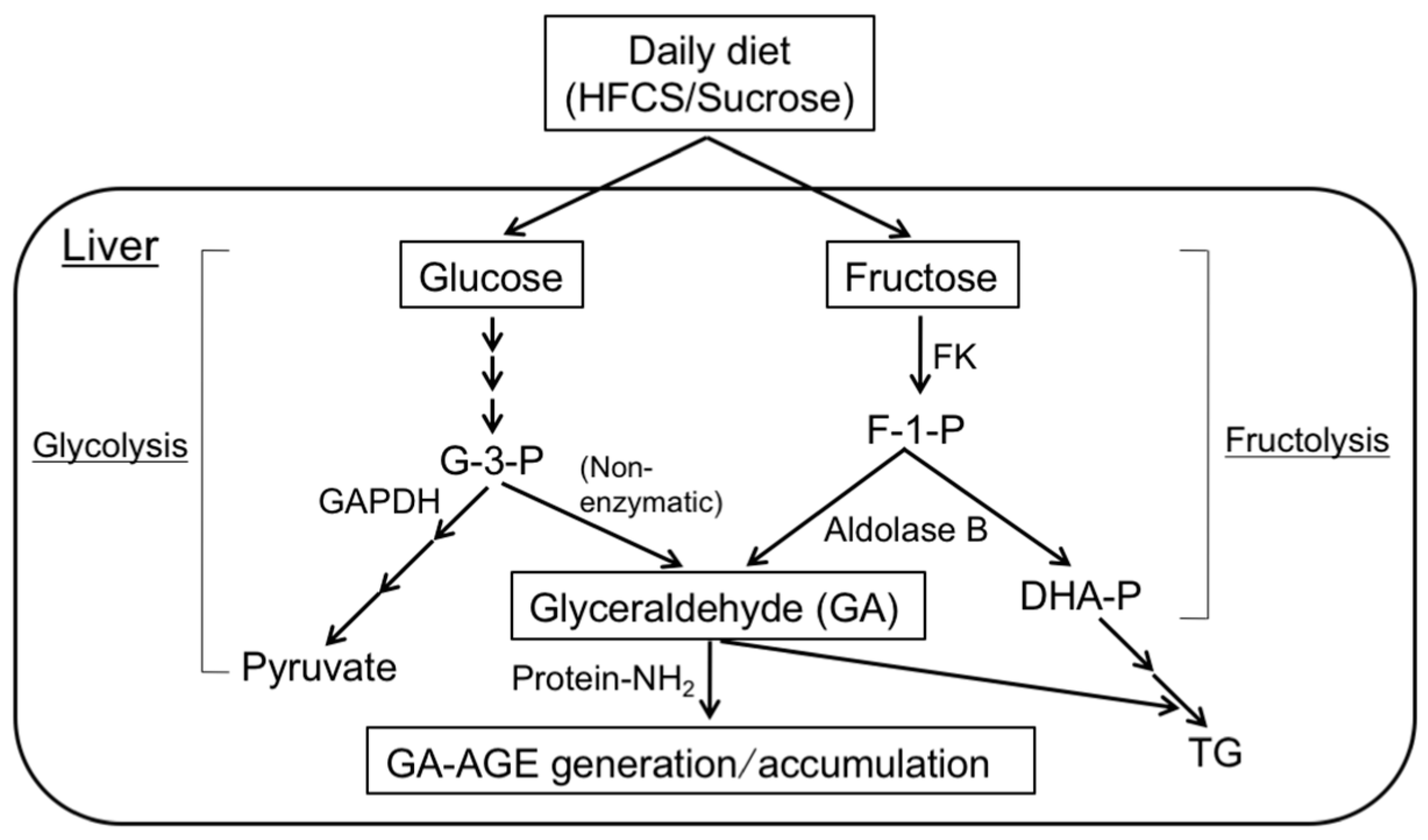
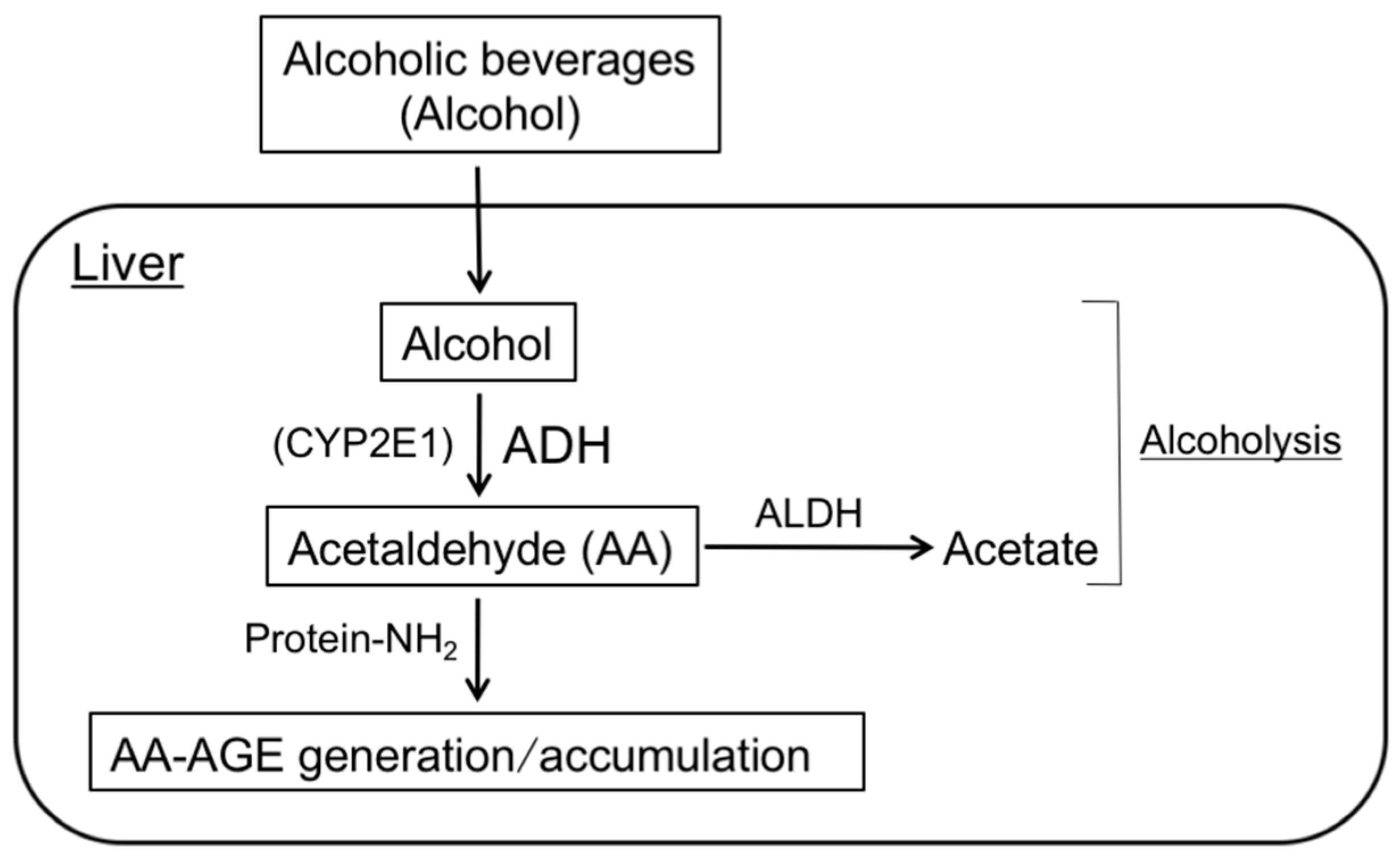
2. Pathway for the Formation of GA- and AA-AGEs in the Liver
3. GA-AGE Theory for the Pathophysiology of NAFLD
3.1. Hepatic IR
3.2. Cytotoxicity of GA-AGEs in Hepatocytes
3.3. Cytotoxicity of GA-AGEs in Hepatic Stellate Cells (HSCs)
3.4. Cytotoxicity of Intracellular GA-AGEs in Hepatocytes
3.5. Intracellular GA-AGE Generation in Fructose
3.6. Serum GA-AGE Levels in NAFL/NASH/HCC
3.7. Serum GA-AGE Levels in CVD
3.8. Neurotoxicity of GA-AGEs
4. AA-AGE Theory for the Pathogenesis of ALD
4.1. Cytotoxicity of AA-AGEs in Hepatocytes
4.2. Cytotoxicity of AA-AGEs in HSCs
4.3. Hepatic AA-AGEs Reflect the Degree of ALD during the Chronic Consumption of Alcohol
4.4. Staining of AA-AGEs in ALD Patients
4.5. Neurotoxicity of AA-AGEs
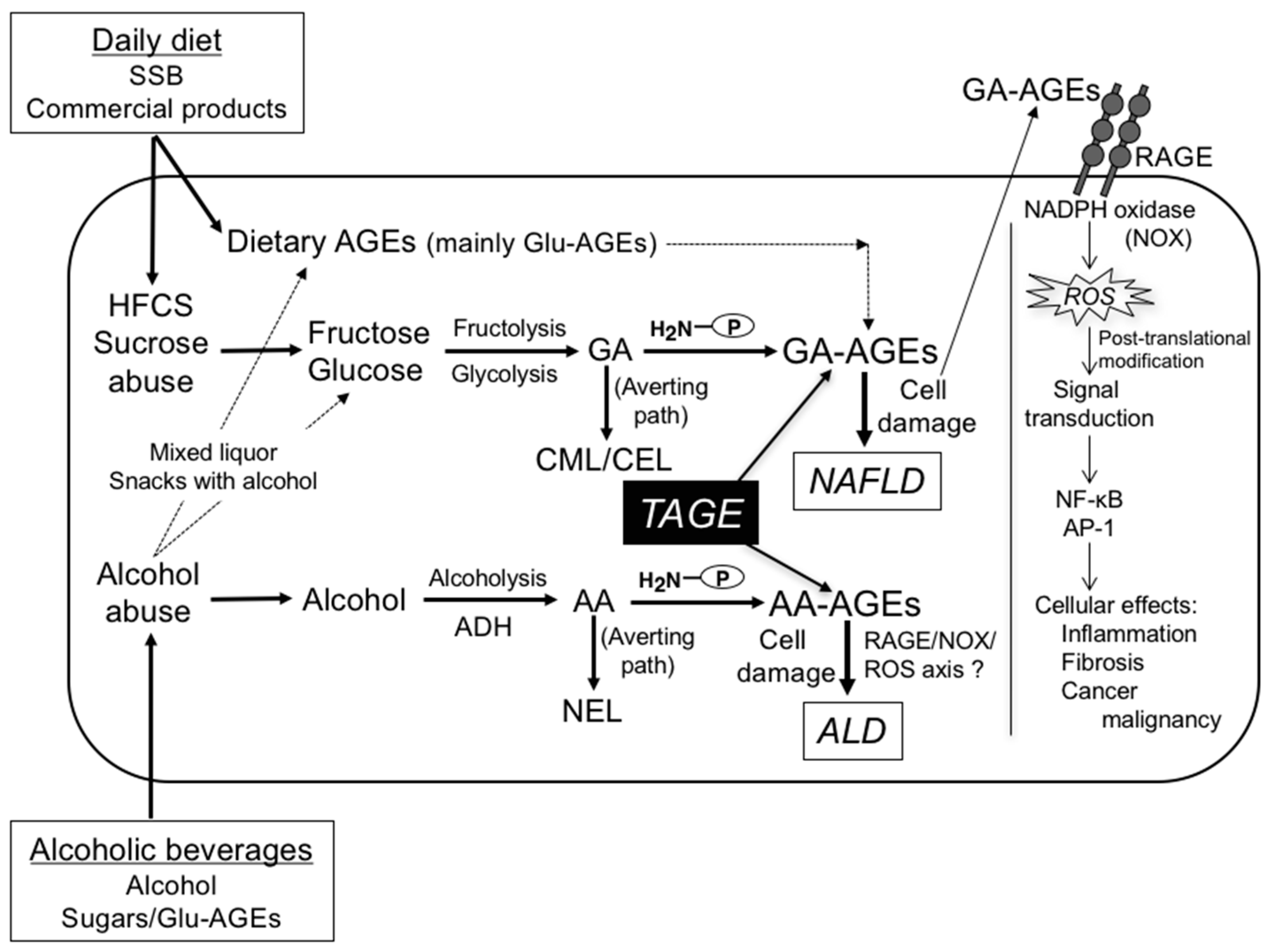
5. Prevention of the Generation/Accumulation of GA- and AA-AGEs in the Liver
5.1. Sugars (HFCS/Sucrose)
5.1.1. GA-AGE Generation/Accumulation
5.1.2. Sugar Content in Soft Drinks and Alcoholic Beverages
5.1.3. Restricting the Consumption of SSB and Alcoholic Beverages
5.2. Dietary Glu-AGEs
5.2.1. GA-AGE Generation/Accumulation
5.2.2. Glu-AGE Contents in Soft Drinks, Foods, and Alcoholic Beverages
5.2.3. Restricting the Consumption of Glu-AGEs
6. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AA | Acetaldehyde |
| AA-AGEs | Acetaldehyde-derived AGEs |
| Aβ | Amyloid beta |
| AD | Alzheimer’s disease |
| ADH | Alcohol dehydrogenase |
| AGEs | Advanced glycation end-products |
| AHA | American Heart Association |
| ALD | Alcoholic liver disease |
| ALDH | Aldehyde dehydrogenase |
| CEL | N-(Carboxyethyl)lysine |
| CML | N-(Carboxymethyl)lysine |
| CRF | Chronic renal failure |
| CRP | C-Reactive protein |
| CSF | Cerebrospinal fluid |
| CVD | Cardiovascular disease |
| CYP2E1 | Cytochrome P450 family 2, subfamily E, polypeptide 1 |
| ECs | Endothelial cells |
| ELISA | Enzyme-linked immunosorbent assay |
| Fru-AGEs | Fructose-derived AGEs |
| GA | Glyceraldehyde |
| GA-AGEs | Glyceraldehyde-derived AGEs |
| GA-3-P | Glyceraldehyde-3-phosphate |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| Glu-AGEs | Glucose-derived AGEs |
| HbA1c | Hemoglobin A1c |
| HCC | Hepatocellular carcinoma |
| HFCS | High-fructose corn syrup |
| 4-HNE | 4-Hydroxy-2-nonenal |
| hnRNPM | Heterogeneous nuclear ribonucleoprotein M |
| Hsc70 | Heat shock cognate 70 |
| HSCs | Hepatic stellate cells |
| IR | Insulin resistance |
| IRS-1 | Insulin receptor substrate-1 |
| LSRD | Lifestyle-related diseases |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MetS | Metabolic syndrome |
| MFB | Myofibroblasts |
| NAFL | Non-alcoholic fatty liver |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NBNC-HCC | Non-B or non-C HCC |
| NEL | N-(Ethyl)lysine |
| NF-κB | Nuclear factor-κB |
| PPAR-γ | Peroxisome proliferator-activated receptor-γ |
| RAGE | Receptor for AGEs |
| ROS | Reactive oxygen species |
| SSB | Sugar-sweetened beverages |
| TAGE | Toxic AGEs |
| T2DM | Type 2 diabetes mellitus |
| TGF-β1 | Transforming growth factor-β1 |
| TNF-α | Tumor necrosis factor-α |
| VEGF | Vascular endothelial growth factor |
| WHO | World Health Organization |
References
- Vuppalanchi, R.; Chalasani, N. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: Selected practical issues in their evaluation and management. Hepatology 2009, 49, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, R.S.; Dasarathy, S.; McCullough, A.J. Practice Guideline Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Alcoholic liver disease. Hepatology 2010, 51, 307–328. [Google Scholar] [PubMed]
- World Health Organization. Alcohol Fact Sheet. WHO Media Center Fact Sheets 2015. Available online: http://www.who.int/mediacentre/factsheets/fs349/en/ (accessed on 6 June 2017).
- Tannapfel, A.; Denk, H.; Dienes, H.P.; Langner, C.; Schirmacher, P.; Trauner, M.; Flott-Rahmel, B. Histopathological diagnosis of non-alcoholic and alcoholic fatty liver disease. Virchows Arch. 2011, 458, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, S. Implications of diet on nonalcoholic fatty liver disease. Curr. Opin. Gastroenterol. 2010, 26, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Dwyer, B.J.; Forbes, S.; Thiel, D.H.; Lewis, P.J.; Ramadori, G. Insulin production and resistance in different models of diet-induced obesity and metabolic syndrome. Int. J. Mol. Sci. 2017, 18, E285. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Basaranoglu, G.; Sabuncu, T.; Sentürk, H. Fructose as a key player in the development of fatty liver disese. World J. Gastroenterol. 2013, 19, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Basaranoglu, G.; Bugianesi, E. Carbohydrate intake and nonalcoholic fatty liver disease: Fructose as a weapon of mass destruction. Hepatobiliary Surg. Nutr. 2015, 4, 109–116. [Google Scholar] [PubMed]
- Jegatheesan, P.; de Bandt, J.P. Fructose and NAFLD: The multifaceted aspects of fructose metabolism. Nutrients 2017, 9, E230. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M.; Nonalcoholic Steatohepatitis Clinical Research Network. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Lazo, M.; Horska, A.; Bonekamp, S.; Lipkin, E.W.; Balasubramanyam, A.; Bantle, J.P.; Johnson, R.J.; Diehl, A.M.; Clark, J.M.; et al. Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology 2012, 56, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A.; Popkin, B.M. Dietary sugar and body weight: Have we reached a crisis in the epidemic of obesity and diabetes?: Health be damned! Pour on the sugar. Diabetes Care 2014, 37, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Lanaspa, M.A.; Rivard, C.J.; Roncal-Jimenez, C.A.; Orlicky, D.J.; Cicerchi, C.; McMahan, R.H.; Abdelmalek, M.F.; Rosen, H.R.; Jackman, M.R.; et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 2013, 58, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Xu, M.; Seyhan, H.A.; Ahmad, S.; Mihm, S.; Ramadori, G.; Schultze, F.C. Diet high infructose leads to an overexpression of lipocalin-2 in rat fatty liver. World J. Gastroenterol. 2014, 20, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Sellmann, C.; Priebs, J.; Landmann, M.; Degen, C.; Engstler, A.J.; Jin, C.J.; Gärttner, S.; Spruss, A.; Huber, O.; Bergheim, I. Diets rich in fructose, fat or fructose and fat alter intestinal barrier function and lead to the development of nonalcoholic fatty liver disease over time. J. Nutr. Biochem. 2015, 26, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; Beutheu, S.; Ventura, G.; Nubret, E.; Sarfati, G.; Bergheim, I.; De Bandt, J.P. Citrulline and nonessential amino acids prevent fructose-induced nonalcoholic fatty liver disease in rats. J. Nutr. 2015, 145, 2273–2279. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; Beutheu, S.; Freese, K.; Waligora-Dupriet, A.J.; Nubret, E.; Butel, M.J.; Bergheim, I.; de Bandt, J.P. Preventive effects of citrulline on western diet-induced non-alcoholic fatty liver disease in rats. Br. J. Nutr. 2016, 116, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Assy, N.; Nasser, G.; Kamayse, I.; Nseir, W.; Beniashvilli, Z.; Djibre, A.; Grosovski, M. Soft drink consumption linked with fatty liver in the absence of traditional risk factors. Can. J. Gastroenterol. 2008, 22, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Abid, A.; Taha, O.; Nseir, W.; Farah, R.; Grosovski, M.; Assy, N. Soft drink consumption is associated with fatty liver disease independent of metabolic syndrome. J. Hepatol. 2009, 51, 918–924. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar]
- Williams, R.; Aspinall, R.; Bellis, M.; Camps-Walsh, G.; Cramp, M.; Dhawan, A.; Ferguson, J.; Forton, D.; Foster, G.; Gilmore, I.; et al. Addressing liver disease in the UK: A blueprint for attaining excellence in health care and reducing premature mortality from lifestyle issues of excess consumption of alcohol, obesity, and viral hepatitis. Lancet 2014, 384, 1953–1997. [Google Scholar] [CrossRef]
- Williams, R.; Ashton, K.; Aspinall, R.; Bellis, M.A.; Bosanquet, J.; Cramp, M.E.; Day, N.; Dhawan, A.; Dillon, J.; Dyson, J.; et al. Implementation of the Lancet Standing Commission on liver disease in the UK. Lancet 2015, 386, 2098–2111. [Google Scholar] [CrossRef]
- Saponaro, C.; Gaggini, M.; Gastaldelli, A. Nonalcoholic fatty liver disease and type 2 diabetes: Common pathophysiologic mechanisms. Curr. Diabetes Rep. 2015, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Doycheva, I.; Cui, J.; Nguyen, P.; Costa, E.A.; Hooker, J.; Hofflich, H.; Bettencourt, R.; Brouha, S.; Sirlin, C.B.; Loomba, R. Non-invasive screening of diabetics in primary care for NAFLD and advanced fibrosis by MRI and MRE. Aliment. Pharmacol. Ther. 2016, 43, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Shen, J.; Sun, T.T.; Zhang, X.; Wong, N. Obesity, insulin resistance, NASH and hepatocellular carcinoma. Semin. Cancer Biol. 2013, 23, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Dyson, J.; Jaques, B.; Chattopadyhay, D.; Lochan, R.; Graham, J.; Das, D.; Aslam, T.; Patanwala, I.; Gaggar, S.; Cole, M.; et al. Hepatocellular cancer: The impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol. 2014, 60, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [PubMed]
- Chitturi, S.; Abeygunasekera, S.; Farrell, G.C.; Holmes-Walker, J.; Hui, J.M.; Fung, C.; Karim, R.; Lin, R.; Samarasinghe, D.; Liddle, C.; et al. NASH and insulin resistance: Insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology 2002, 35, 373–379. [Google Scholar] [CrossRef] [PubMed]
- NIAAA. Alcohol Facts and Statistics. Available online: http://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/alcohol-facts-and-statistics (accessed on 6 June 2017).
- Bellentani, S.; Saccoccio, G.; Costa, G.; Tiribelli, C.; Manenti, F.; Sodde, M.; Saveria Crocè, L.; Sasso, F.; Pozzato, G.; Cristianini, G.; et al. Drinking habits as cofactors of risk for alcohol induced liver damage. Gut 1997, 41, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Becker, U.; Deis, A.; Sørensen, T.I.; Grønbaek, M.; Borch-Johnsen, K.; Müller, C.F.; Schnohr, P.; Jensen, G. Prediction of risk of liver disease by alcohol intake, sex, and age: A prospective population study. Hepatology 1996, 23, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Naveau, S.; Giraud, V.; Borotto, E.; Aubert, A.; Capron, F.; Chaput, J.C. Excess weight risk factor for alcoholic liver disease. Hepatology 1997, 25, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Kerr, W.C.; Fillmore, K.M.; Marvy, P. Beverage-specific alcohol consumption and cirrhosis mortality in a group of English-speaking beer-drinking countries. Addiction 2000, 95, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Hatton, J.; Burton, A.; Nash, H.; Munn, E.; Burgoyne, L.; Sheron, N. Drinking patterns, dependency and life-time drinking history in alcohol-related liver disease. Addiction 2009, 104, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Bucala, R.; Cerami, A. Advanced glycosylation: Chemistry, biology, and implications for diabetes and aging. Adv. Pharmacol. 1992, 23, 1–34. [Google Scholar] [PubMed]
- Vlassara, H.; Bucala, R.; Striker, L. Pathogenic effects of advanced glycosylation: Biochemical, biologic, and clinical implications for diabetes and aging. Lab. Invest. 1994, 70, 138–151. [Google Scholar] [PubMed]
- Brownlee, M. Advanced protein glycosylation in diabetes and aging. Ann. Rev. Med. 1995, 46, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Makita, Z. Alternative routes for the formation of immunochemically distinct advanced glycation end-products in vivo. Curr. Mol. Med. 2001, 1, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Iwaki, M.; Shimogaito, N.; Wu, X.; Yamagishi, S.; Takeuchi, M. TAGE (toxic AGEs) theory in diabetic complications. Curr. Mol. Med. 2006, 6, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Yamagishi, S. Involvement of toxic AGEs (TAGE) in the pathogenesis of diabetic vascular complications and Alzheimer’s disease. J. Alzheimers Dis. 2009, 16, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Takino, J.; Yamagishi, S. Involvement of the toxic AGEs (TAGE)-RAGE system in the pathogenesis of diabetic vascular complications: A novel therapeutic strategy. Curr. Drug Targets 2010, 11, 1468–1482. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Yamagishi, S. TAGE (toxic AGEs) hypothesis in various chronic diseases. Med. Hypotheses 2004, 63, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Hyogo, H.; Yamagishi, S.; Iwamoto, K.; Arihiro, K.; Takeuchi, M.; Sato, T.; Ochi, H.; Nonaka, M.; Nabeshima, Y.; Inoue, M.; et al. Elevated levels of serum advanced glycation end products in patients with non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2007, 22, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, K.; Kanno, K.; Hyogo, H.; Yamagishi, S.; Takeuchi, M.; Tazuma, S.; Chayama, K. Advanced glycation end products enhance the proliferation and activation of hepatic stellate cells. J. Gastroenterol. 2008, 43, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Hyogo, H.; Yamagishi, S. Advanced glycation end products (AGEs) and their involvement in liver disease. Curr. Pharm. Des. 2008, 14, 969–972. [Google Scholar] [CrossRef] [PubMed]
- Hyogo, H.; Chayama, K.; Yamagishi, S. Nonalcoholic fatty liver disease and cardiovascular disease. Curr. Pharm. Des. 2014, 20, 2403–2411. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Bucala, R.; Suzuki, T.; Ohkubo, T.; Yamazaki, M.; Koike, T.; Kameda, Y.; Makita, Z. Neurotoxicity of advanced glycation end-products for cultured cortical neurons. J. Neuropathol. Exp. Neurol. 2000, 59, 1094–1105. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Watai, T.; Sasaki, N.; Choei, H.; Iwaki, M.; Ashizawa, T.; Inagaki, Y.; Yamagishi, S.; Kikuchi, S.; Riederer, P.; et al. Neurotoxicity of acetaldehyde-derived advanced glycation end products for cultured cortical neurons. J. Neuropathol. Exp. Neurol. 2003, 62, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, N.; George, J.; Takeuchi, M.; Fukumura, A.; Toshikuni, N.; Arisawa, T.; Tsutsumi, M. Acetaldehyde-derived advanced glycation end-products promote alcoholic liver diasease. PLoS ONE 2013, 8, e70034. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, M.; Adachi, H.; Yamagishi, S.; Takeuchi, M.; Furuki, K.; Hino, A.; Hiratsuka, A.; Takajo, Y.; Imaizumi, T. Positive association of serum levels of advanced glycation end products with thrombogenic markers in humans. Metabolism 2006, 55, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Adachi, H.; Takeuchi, M.; Enomoto, M.; Furuki, K.; Matsui, T.; Nakamura, K.; Imaizumi, T. Serum level of advanced glycation end-products (AGEs) is an independent determinant of plasminogen activator inhibitor-1 (PAI-1) in nondiabetic general population. Horm. Metab. Res. 2007, 39, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Tahara, N.; Yamagishi, S.; Kodama, N.; Tahara, A.; Honda, A.; Nitta, Y.; Igata, S.; Matsui, T.; Takeuchi, M.; Kaida, H.; et al. Clinical and biochemical factors associated with area and metabolic activity in the visceral and subcutaneous adipose tissues by FDG-PET/CT. J. Clin. Endocrinol. Metab. 2015, 100, E739–E747. [Google Scholar] [CrossRef] [PubMed]
- Kajikawa, M.; Nakashima, A.; Fujimura, N.; Maruhashi, T.; Iwamoto, Y.; Iwamoto, A.; Matsumoto, T.; Oda, N.; Hidaka, T.; Kihara, Y.; et al. Ratio of serum levels of AGEs to soluble form of RAGE is a predictor of endothelial function. Diabetes Care 2015, 38, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, M.; Oyama, J.; Takeuchi, M.; Shibata, Y.; Yamamoto, Y.; Kawasaki, T.; Komoda, H.; Kodama, K.; Sakuma, M.; Toyoda, S.; et al. Acute effects of statin on reduction of angiopoietin-like 2 and glyceraldehyde-derived advanced glycation end-products levels in patients with acute myocardial infarction: A message from SAMIT (Statin for Acute Myocardial Infarction Trial). Heart Vessel. 2016, 31, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Takino, J.; Yamagishi, S. Involvement of TAGE-RAGE system in the pathogenesis of diabetic retinopathy. J. Ophthalmol. 2010, 2010, 170393. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Kikuchi, S.; Sasaki, N.; Suzuki, T.; Watai, T.; Iwaki, M.; Bucala, R.; Yamagishi, S. Involvement of advanced glycation end-products (AGEs) in Alzheimer’s disease. Curr. Alzheimer Res. 2004, 1, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Choei, H.; Sasaki, N.; Takeuchi, M.; Yoshida, T.; Ukai, W.; Yamagishi, S.; Kikuchi, S.; Saito, T. Glyceraldehyde-derived advanced glycation end products in Alzheimer’s disease. Acta Neuropathol. 2004, 108, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Shimogaito, N.; Wu, X.; Kikuchi, S.; Yamagishi, S.; Takeuchi, M. Toxic advanced glycation end product (TAGE) theory in Alzheimer’s disease. Am. J. Alzheimers Dis. Other Demen. 2006, 21, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Yamagishi, S. Possible involvement of advanced glycation end-products (AGEs) in the pathogenesis of Alzheimer’s disease. Curr. Pharm. Des. 2008, 14, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Shimizu, T.; Sugawara, H.; Watanabe, H.; Nakamura, H.; Choei, H.; Sasaki, N.; Yamagishi, S.; Takeuchi, M.; Shimizu, H. Regulation of human melanoma growth and metastasis by AGE-AGE receptor interactions. J. Invest. Dermatol. 2004, 122, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Yamagishi, S. AGE-RAGE system and carcinogenesis. Curr. Pharm. Des. 2008, 14, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Takino, J.; Yamagishi, S.; Takeuchi, M. Cancer malignancy is enhanced by glyceraldehyde-derived advanced glycation end-products. J. Oncol. 2010, 2010, 739852. [Google Scholar] [CrossRef] [PubMed]
- Takino, J.; Yamagishi, S.; Takeuchi, M. Glycer-AGEs-RAGE signaling enhances the angiogenic potential of hepatocellular carcinoma by upregulating VEGF expression. World J. Gastroenterol. 2012, 18, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.Y.; Takeuchi, M.; Hyogo, H.; McKeown-Eyssen, G.; Yamagishi, S.; Chayama, K.; O'Brien, P.J.; Ferrari, P.; Overvad, K.; Olsen, A.; et al. The association between glyceraldehyde-derived advanced glycation end-products and colorectal cancer risk. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1855–1863. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, C.G.; Stehouwer, C.D.; van Hinsbergh, V.W. Fructose-mediated non-enzymatic glycation: Sweet coupling or bad modification. Diabetes Metab. Res. Rev. 2004, 20, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Gaby, A.R. Adverse effects of dietary fructose. Altern. Med. Rev. 2005, 10, 294–306. [Google Scholar] [PubMed]
- Hallfrisch, J. Metabolic effects of dietary fructose. FASEB J. 1990, 4, 2652–2660. [Google Scholar] [PubMed]
- Mayes, P.A. Intermediary metabolism of fructose. Am. J. Clin. Nutr. 1993, 58, 754S–765S. [Google Scholar] [PubMed]
- Takeuchi, M.; Yamagishi, S. Alternative routes for the formation of glyceraldehyde-derived AGEs (TAGE) in vivo. Med. Hypotheses 2004, 63, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Alcoholic fatty liver: Its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 2004, 34, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, M.W.; Hedegaard, R.V.; Andersen, J.M.; de Courten, B.; Bügel, S.; Nielsen, J.; Skibsted, L.H.; Dragsted, L.O. Advanced glycation endproducts in food and their effects on health. Food Chem. Toxicol. 2013, 60, 10–37. [Google Scholar] [CrossRef] [PubMed]
- Dankner, R.; Chetrit, A.; Shanik, M.H.; Raz, I.; Roth, J. Basal-state hyperinsulinemia in healthy normoglycemic adults is predictive of type 2 diabetes over a 24-year follow-up: A preliminary report. Diabetes Care 2009, 32, 1464–1466. [Google Scholar] [CrossRef] [PubMed]
- Kasai, T.; Miyauchi, K.; Kajimoto, K.; Kubota, N.; Dohi, T.; Kurata, T.; Amano, A.; Daida, H. The adverse prognostic significance of the metabolic syndrome with and without hypertension in patients who underwent complete coronary revascularization. J. Hypertens. 2009, 27, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Agnoli, C.; Berrino, F.; Abagnato, C.A.; Muti, P.; Panico, S.; Crosignani, P.; Krogh, V. Metabolic syndrome and postmenopausal breast cancer in the ORDET cohort: A nested case-control study. Nutr. Metab. Cardiovasc. Dis. 2010, 20, 41–48. [Google Scholar] [CrossRef] [PubMed]
- García, R.G.; Rincόn, M.Y.; Arenas, W.D.; Silva, S.Y.; Reyes, L.M.; Ruiz, S.L.; Ramirez, F.; Camacho, P.A.; Luengas, C.; Saaibi, J.F.; et al. Hyperinsulinemia is a predictor of new cardiovascular events in Colombian patients with a first myocardial infarction. Int. J. Cardiol. 2011, 148, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Faulds, M.H.; Dahlman-Wright, K. Metabolic diseases and cancer risk. Curr. Opin. Oncol. 2012, 24, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Colonna, S.V.; Douglas Case, L.; Lawrence, J.A. A retrospective review of the metabolic syndrome in women diagnosed with breast cancer and correlation with estrogen receptor. Breast Cancer Res. Treat. 2012, 131, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Jinnouchi, Y.; Yamagishi, S.; Takeuchi, M.; Ishida, S.; Jinnouchi, Y.; Jinnouchi, J.; Imaizumi, T. Atorvastatin decreases serum levels of advanced glycation end products (AGEs) in patients with type 2 diabetes. Clin. Exp. Med. 2006, 6, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Tahara, N.; Imaizumi, T.; Takeuchi, M.; Yamagishi, S. Insulin resistance is an independent correlate of high serum levels of advanced glycation end products (AGEs) and low testosterone in non-diabetic men. Oxid. Med. Cell. Longev. 2010, 3, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Tahara, N.; Yamagishi, S.; Matsui, T.; Takeuchi, M.; Nitta, Y.; Kodama, N.; Mizoguchi, M.; Imaizumi, T. Serum levels of advanced glycation end products (AGEs) are independent correlates of insulin resistance in nondiabetic subjects. Cardiovasc. Ther. 2012, 30, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Tahara, N.; Yamagishi, S.; Tahara, A.; Ishibashi, M.; Hayabuchi, N.; Takeuchi, M.; Imaizumi, T. Adiponectin is inversely associated with ratio of serum levels of AGEs to sRAGE and vascular inflammation. Int. J. Cardiol. 2012, 158, 461–462. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Hyogo, H.; Yamagishi, S.; Takeuchi, M.; Ishitobi, T.; Nabeshima, Y.; Arihiro, K.; Chayama, K. Atorvastatin decreases serum levels of advanced glycation endproducts (AGEs) in nonalcoholic steatohepatitis (NASH) patients with dyslipidemia: Clinical usefulness of AGEs as a biomarker for the attenuation of NASH. J. Gastroenterol. 2010, 45, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Yamagishi, S.; Nakamura, K.; Matsui, T.; Imaizumi, T.; Takeuchi, M.; Koga, H.; Ueno, T.; Sara, M. Telmisartan inhibits AGE-induced C-reactive protein production through downregulation of the receptor for AGE via peroxisome proliferator-activated receptor-gamma activation. Diabetologia 2006, 49, 3094–3099. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Yamagishi, S.; Matsui, T.; Nakamura, K.; Ueno, T.; Takeuchi, M.; Sata, M. Telmisartan, an angiotensin II type 1 receptor blocker, inhibits advanced glycation end-product (AGE)-elicited hepatic insulin resistance via peroxisome proliferator-activated receptor-gamma activation. J. Int. Med. Res. 2008, 36, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Yamagishi, S.; Nakamura, K.; Matsui, T.; Imaizumi, T.; Takeuchi, M.; Koga, H.; Ueno, T.; Sata, M. Pigment epithelium-derived factor (PEDF) ameliorates advanced glycation end product (AGE)-induced hepatic insulin resistance in vitro by suppressing Rac-1 activation. Horm. Metab. Res. 2008, 40, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Kitahara, Y.; Kajioka, T.; Takeuchi, M.; Yamagishi, S. Combination therapy with nateglinide and telmisartan ameliorates insulin resistance in Zucker fatty rats by suppressing advanced glycation end product receptor axis. Horm. Metab. Res. 2011, 43, 226–228. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Yamagishi, S.; Nakamura, K.; Matsui, T.; Imaizumi, T.; Takeuchi, M.; Ueno, T.; Sata, M. Pigment epithelium-derived factor (PEDF) inhibits advanced glycation end product (AGE)-induced C-reactive protein expression in hepatoma cells by suppressing Rac-1 activation. FEBS Lett. 2006, 580, 2788–2796. [Google Scholar] [CrossRef] [PubMed]
- Sakuraoka, Y.; Sawada, T.; Okada, T.; Shiraki, T.; Miura, Y.; Hiraishi, K.; Ohsawa, T.; Adachi, M.; Takino, J.; Takeuchi, M.; et al. MK615 decreases RAGE expression and inhibits TAGE-induced proliferation in hepatocellular carcinoma cells. World J. Gastroenterol. 2010, 16, 5334–5341. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Wu, X.; Shimogaito, N.; Takino, J.; Yamagishi, S.; Takeuchi, M. Effects of high-AGE beverage on RAGE and VEGF expressions in the liver and kidneys. Eur. J. Nutr. 2009, 48, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Yamagishi, S.; Inagaki, Y.; Amano, S.; Koga, K.; Abe, R.; Takeuchi, M.; Ohno, S.; Yoshimura, A.; Makita, Z. Angiogenesis induced by advanced glycation end products and its prevention by cerivastatin. FASEB J. 2002, 16, 1928–1930. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T.; Nakamura, K.; Inoue, H.; Takeuchi, M.; Ueda, S.; Fukami, K.; Okuda, S.; Imaizumi, T. Olmesartan blocks advanced glycation end products (AGEs)-induced angiogenesis in vitro by suppressing receptor for AGEs (RAGE) expression. Microvasc. Res. 2008, 75, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Nakamura, K.; Matsui, T.; Yoshida, T.; Takeuchi, M.; Imaizumi, T. Pigment epithelium-derived factor (PEDF) blocks advanced glycation end product (AGE)-induced angiogenesis in vitro. Horm. Metab. Res. 2007, 39, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Nakamura, K.; Matsui, T.; Inagaki, Y.; Takenaka, K.; Jinnouchi, Y.; Yoshida, Y.; Matsuura, T.; Narama, I.; Motomiya, Y.; et al. Pigment epithelium-derived factor inhibits advanced glycation end product-induced retinal vascular hyperpermeability by blocking reactive oxygen species-mediated vascular endothelial growth factor expression. J. Biol. Chem. 2006, 281, 20213–20220. [Google Scholar] [CrossRef] [PubMed]
- Niiya, Y.; Abumiya, T.; Yamagishi, S.; Takino, J.; Takeuchi, M. Advanced glycation end products increase permeability of brain microvascular endothelial cells through reactive oxygen species-induced vascular endothelial growth factor expression. J. Stroke Cerebrovasc. Dis. 2012, 21, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T.; Nakamura, K.; Inoue, H.; Takeuchi, M.; Ueda, S.; Okuda, S.; Imaizumi, T. Olmesartan blocks inflammatory reactions in endothelial cells evoked by advanced glycation end products by suppressing generation of reactive oxygen species. Ophthalmic. Res. 2008, 40, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Niiya, Y.; Abumiya, T.; Shichinohe, H.; Kuroda, S.; Kikuchi, S.; Ieko, M.; Yamagishi, S.; Takeuchi, M.; Sato, T.; Iwasaki, Y. Susceptibility of brain microvascular endothelial cells to advanced glycation end products-induced tissue factor upregulation is associated with intracellular reactive oxygen species. Brain Res. 2006, 1108, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, Y.; Yamagishi, S.; Okamoto, T.; Takeuchi, M.; Amano, S. Pigment epithelium-derived factor prevents advanced glycation end products-induced monocyte chemoattractant protein-1 production in microvascular endothelial cells by suppressing intracellular reactive oxygen species generation. Diabetologia 2003, 46, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbach, H.; Weiskirchen, R.; Kasper, M.; Gressner, A.M. Up-regulated expression of the receptor for advanced glycation end products in cultured rat hepatic stellate cells during transdifferentiation to myofibroblasts. Hepatology 2001, 34, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Takino, J.; Kobayashi, Y.; Takeuchi, M. The formation of intracellular glyceraldehyde-derived advanced glycation end-products and cytotoxicity. J. Gastroenterol. 2010, 45, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Ebata, Y.; Takino, J.; Tsuchiya, H.; Sakabe, T.; Ikeda, Y.; Hama, S.; Kogure, K.; Takeuchi, M.; Shiota, G. Presence of glyceraldehyde-derived advanced glycation end-products in the liver of insulin-resistant mice. Int. J. Vitam. Nutr. Res. 2013, 83, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Takino, J.; Nagamine, K.; Takeuchi, M.; Hori, T. In vitro identification of nonalcoholic fatty liver disease-related protein hnRNPM. World J. Gastroenterol. 2015, 21, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Vilà, L.; Rebollo, A.; Ađalsteisson, G.S.; Alegret, M.; Merlos, M.; Roglans, N.; Laguna, J.C. Reduction of liver fructokinase expression and improved hepatic inflammation and metabolism in liquid fructose-fed rats after atorvastatin treatment. Toxicol. Appl. Pharmacol. 2011, 251, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Takino, J.; Sakasai-Sakai, A.; Takata, T.; Ueda, T.; Tsutsumi, M.; Hyogo, H.; Yamagishi, S. Involvement of the TAGE-RAGE system in non-alcoholic steatohepatitis: Novel treatment strategies. World J. Hepatol. 2014, 6, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Sakasai-Sakai, A.; Takino, J.; Takata, T.; Ueda, T.; Tsutsumi, M. Toxic AGEs (TAGE) theory in the pathogenesis of NAFLD and ALD. Int. J. Diabetes Clin. Res. 2015, 2, 4. [Google Scholar] [CrossRef]
- Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Ueda, T.; Takino, J.; Tsutsumi, M.; Hyogo, H.; Yamagishi, S. Serum levels of toxic AGEs (TAGE) may be a promising novel biomarker in development and progression of NASH. Med. Hypotheses 2015, 84, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Takino, J.; Nagamine, K.; Hori, T.; Sakasai-Sakai, A.; Takeuchi, M. Contribution of the toxic advanced glycation end-products-receptor axis in nonalcoholic steatohepatitis-related hepatocellular carcinoma. World J. Hepatol. 2015, 7, 2459–2469. [Google Scholar] [CrossRef] [PubMed]
- Kan, H.; Yamagishi, S.; Ojima, A.; Fukami, K.; Ueda, S.; Takeuchi, M.; Hyogo, H.; Aikata, H.; Chayama, K. Elevation of serum levels of advanced glycation end products in patients with non-B or non-C hepatocellular carcinoma. J. Clin. Lab. Anal. 2015, 29, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Scorletti, E.; Calder, P.C.; Byrne, C.D. Non-alcoholic fatty liver disease and cardiovascular risk: Metabolic aspects and novel treatments. Endocrine 2011, 40, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Fargion, S.; Porzio, M.; Fracanzani, A.L. Nonalcoholic fatty liver disease and vascular disease: State-of-art. World J. Gastroenterol. 2014, 20, 13306–13324. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, I.; Rinella, M.E. NAFLD and cardiovascular disease: Can the real association be determined? Curr. Hepatol. Rep. 2014, 13, 130–141. [Google Scholar] [CrossRef]
- Tahara, N.; Yamagishi, S.; Takeuchi, M.; Honda, A.; Tahara, A.; Nitta, Y.; Kodama, N.; Mizoguchi, M.; Kaida, H.; Ishibashi, M.; et al. Positive association between serum level of glyceraldehyde-derived advanced glycation end products and vascular inflammation evaluated by [18F]-fluorodeoxyglucose positron emission tomography. Diabetes Care 2012, 35, 2618–2625. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Yamagishi, S.; Matsui, T.; Noda, Y.; Ueda, S.; Jinnouchi, Y.; Sasaki, K.; Takeuchi, M.; Imaizumi, T. Serum levels of advanced glycation end products (AGEs) are inversely associated with the number and migratory activity of circulating endothelial progenitor cells in apparently healthy subjects. Cardiovasc. Ther. 2012, 30, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, Y.; Daida, H.; Morimoto, T.; Kasai, T.; Miyauchi, K.; Yamagishi, S.; Takeuchi, M.; Hiro, T.; Kimura, T.; Nakagawa, Y.; et al. Relationship between advanced glycation end products and plaque progression in patients with acute coronary syndrome: The JAPAN-ACS sub-study. Cardiovasc. Diabetol. 2013, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Kawai, Y.; Kitayama, M.; Akao, H.; Motoyama, A.; Wakasa, M.; Saito, R.; Aoki, H.; Fujibayashi, K.; Tsuchiya, T.; et al. Diurnal glycemic fluctuation is associated with severity of coronary artery disease in prediabetic patients: Possible role of nitrotyrosine and glyceraldehyde-derived advanced glycation end products. J. Cardiol. 2017, 69, 625–631. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Tong, M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Ryoo, N.Y.; Shin, D.W.; Trojanowski, J.Q.; Shaw, L.M. Role of cerebrospinal fluid biomarkers in clinical trials for Alzheimer’s disease modifying therapies. Korean J. Physiol. Pharmacol. 2014, 18, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, E.; Issa, R.; Sjögren, M.; Wallin, A.; Blennow, K.; Tarkowski, A.; Kumar, P. Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer’s disease and vascular dementia. Neurobiol. Aging 2002, 23, 237–243. [Google Scholar] [CrossRef]
- Zetterberg, H.; Andreasen, N.; Blennow, K. Increased cerebrospinal fluid levels of transforming growth factor-β1 in Alzheimer’s disease. Neurosci. Lett. 2004, 367, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Koriyama, Y.; Furukawa, A.; Muramatsu, M.; Takino, J.; Takeuchi, M. Glyceraldehyde caused Alzheimer’s disease-like alterations in diagnostic marker levels in SH-SY5Y human neuroblastoma cells. Sci. Rep. 2015, 5, 13313. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Sato, T.; Takino, J.; Kobayashi, Y.; Furuno, S.; Kikuchi, S.; Yamagishi, S. Diagnostic utility of serum or cerebrospinal fluid levels of toxic advanced glycation end-products (TAGE) in early detection of Alzheimer’s disease. Med. Hypotheses 2007, 69, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Hirose, D.; Hanyu, H.; Fukusawa, R.; Hatanaka, H.; Namioka, N.; Okita, M. Circulating levels of advanced glycation end products in diabetes mellitus-related dementia. J. Am. Geriatr. Soc. 2015, 63, 2196–2198. [Google Scholar] [CrossRef] [PubMed]
- Beier, J.I.; McClain, C.J. Mechanisms and cell signaling in alcoholic liver disease. Biol. Chem. 2010, 391, 1249–1264. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Setshedi, M.; Wands, J.R.; Monte, S.M. Acetaldehyde adducts in alcoholic liver disease. Oxid. Med. Cell. Longev. 2010, 3, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Upadhya, S.C.; Ravindranath, V. Detection and localization of protein acetaldehyde adducts in rat brain after chronic ethanol treatment. Alcohol. Clin. Exp. Res. 2002, 26, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Mucci, L.; Santilli, F.; Cuccurullo, C.; Davi, G. Cardiovascular risk and dietary sugar intake: Is the link so sweet? Intern. Emerg. Med. 2012, 7, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.S.; Hu, F.B. Fructose and cardiometabolic health: What the evidence from sugar-sweetened beverages tells us. J. Am. Coll. Cardiol. 2015, 66, 1615–1624. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Fox, C.S.; Jacques, P.F.; Speliotes, E.K.; Hoffmann, U.; Smith, C.E.; Saltzman, E.; McKeown, N.M. Sugar-sweetened beverage, diet soda, and fatty liver disease in the Framingham heart study cohorts. J. Hepatol. 2015, 63, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Wehmeyer, M.H.; Zyriax, B.C.; Jagemann, B.; Roth, E.; Windler, E.; ZurWiesch, J.S.; Lohse, A.W.; Kluwe, J. Nonalcoholic fatty liver disease is associated with excessive calorie intake rather than a distinctive dietary pattern. Medicine 2016, 95, e3887. [Google Scholar] [CrossRef] [PubMed]
- Rippe, J.M.; Angelopoulos, T.J. Sugars, obesity, and cardiovascular disease: Results from recent randomized control trials. Eur. J. Nutr. 2016, 55, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M. Serum levels of toxic AGEs (TAGE) may be a promising novel biomarker for the onset/progression of lifestyle-related diseases. Diagnostics 2016, 6, E23. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Invest. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.; Bremer, A.A.; Lustig, R.H. What is metabolic syndrome, and why are children getting it? Ann. N. Y. Acad. Sci. 2013, 1281, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Schwarz, J.M.; Havel, P.J. Adverse metabolic effects of dietary fructose: Results from the recent epidemiological, clinical, and mechanistic studies. Curr. Opin. Lipidol. 2013, 24, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Noworolski, S.M.; Wen, M.J.; Dyachenko, A.; Pior, J.L.; Weinberg, M.E.; Herraiz, L.A.; Tai, V.W.; Bergeron, N.; Bersot, T.P.; et al. Effect of a high-fructose weight maintaining diet on lipogenesis and liver fat. J. Clin. Endocrinol. Metab. 2015, 100, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.H.; Mulligan, K.; Noworolski, S.M.; Tai, V.W.; Wen, M.J.; Erkin-Cakmak, A.; Gugliucci, A.; Schwarz, J.M. Isocaloric fructose restriction and metabolic improvement in children with obesity and metabolic syndrome. Obesity 2016, 24, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Gebhardt, R. Dietary fructose as a risk factor for non-alcoholic fatty liver disease (NAFLD). Arch. Toxicol. 2017, 91, 1545–1563. [Google Scholar] [CrossRef] [PubMed]
- Joint World Health Organization (WHO)/Food and Agriculture Organization (FAO) Expert Consultation. Diet, Nutrition and the Prevention of Chronic Diseases; World Health Organization (WHO) Technical Report Series; World Health Organization: Geneva, Switzerland, 2003; p. 916. [Google Scholar]
- Lustig, R.H. Fructose: Metabolic, hedonic, and societal parallels with ethanol. J. Am. Diet. Assoc. 2010, 110, 1307–1321. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Lê, K.A.; Tran, C.; Paquot, N. Fructose and metabolic diseases: New findings, new questions. Nutrition 2010, 26, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Xu, M.; Schultze, F.C.; Wilting, J.; Mihm, S.; Raddatz, D.; Ramadori, G. Combination of alcohol and fructose exacerbates metabolic imbalance in terms of hepatic damage, dyslipidemia, and insulin resistance in rats. PLoS ONE 2014, 9, e104220. [Google Scholar] [CrossRef] [PubMed]
- Jonson, R.K.; Appel, L.J.; Brands, M.; Howard, B.V.; Lefevre, M.; Lustig, R.H.; Sacks, F.; Steffen, L.M.; Wylie-Rosett, J.; American Heart Association Nutrition Committee of the Council on Nutrition; et al. Dietary sugars intake and cardiovascular health: A scientific statement from the American Heart Association. Circulation 2009, 120, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Sugars Intake for Adults and Children. Available online: http://apps.who.int/iris/bitstream/10665/149782/1/9789241549028_eng.pdf?ua=1 (accessed on 6 June 2017).
- Takeuchi, M.; Takino, J.; Shirai, H.; Kawakami, M.; Furuno, S.; Kobayashi, Y. Assessment of total sugar and glucose concentrations in commonly consumed beverages in Japan. Nutr. Food Technol. 2015, 1, 2. [Google Scholar]
- Imamura, F.; O’Connor, L.; Ye, Z.; Mursu, J.; Hayashino, Y.; Bhupathiraju, S.N.; Forouhi, N.G. Consumption of sugar sweetened beverages, artificially sweetened beverages, and fruit juice and incidence of type 2 diabetes: Systematic review, meta-analysis, and estimation of population attributable fraction. BMJ 2015, 351, h3576. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Takino, J.; Furuno, S.; Shirai, H.; Kawakami, M.; Muramatsu, M.; Kobayashi, Y.; Yamagishi, S. Assessment of the concentrations of various advanced glycation end-products in beverages and foods that are commonly consumed in Japan. PLoS ONE 2015, 10, e0118652. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Striker, G.E. AGE restriction in diabetes mellitus: A paradigm shift. Nat. Rev. Endocrinol. 2011, 7, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Kellow, N.J.; Savige, G.S. Dietary advanced glycation end-product restriction for the attenuation of insulin resistance, oxidative stress and endothelial dysfunction: A systematic review. Eur. J. Clin. Nutr. 2013, 67, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Gebauer, S.K.; Baer, D.J.; Sun, K.; Turner, R.; Silber, H.A.; Talegawkar, S.; Ferrucci, L.; Novotny, J.A. Dietary intake of advanced glycation end products did not affect endothelial function and inflammation in healthy adults in a randomized controlled trial. J. Nutr. 2014, 44, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Palimeri, S.; Palioura, E.; Diamanti-Kandarakis, E. Current perspectives on the health risks associated with the consumption of advanced glycation end products: Recommendations for dietary management. Diabetes Metab. Syndr. Obes. 2015, 8, 415–426. [Google Scholar] [PubMed]
- Aragno, M.; Mastrocola, R. Dietary sugars and endogenous formation of advanced glycation endproducts: Emerging mechanisms of disease. Nutrients 2017, 9, 385. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Yamagishi, S.; Takeuchi, M.; Kohno, K.; Shibata, R.; Matsumoto, Y.; Kaneyuki, U.; Fujimura, T.; Hayashida, A.; Okuda, S. Oral adsorbent AST-120 decreases serum levels of AGEs in patients with chronic renal failure. Mol. Med. 2006, 12, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Kellow, N.J.; Coughlan, M.T. Effect of diet-derived advanced glycation end products on inflammation. Nutr. Rev. 2015, 73, 737–759. [Google Scholar] [CrossRef] [PubMed]
- De Courten, B.; de Courten, M.P.; Soldatos, G.; Dougherty, S.L.; Straznicky, N.; Schlaich, M.; Sourris, K.C.; Chand, V.; Scheijen, J.L.; Kingwell, B.A.; et al. Diet low in advanced glycation end products increases insulin sensitivity in healthy overweight individuals: A double-blind, randomized, crossover trial. Am. J. Clin. Nutr. 2016, 103, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.E.; Dordevic, A.L.; Tan, S.M.; Ryan, L.; Coughlan, M.T. Dietary advanced glycation end products and risk factors for chronic disease: A systematic review of randomised controlled trials. Nutrients 2016, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Makita, Z.; Yanagisawa, K.; Kamaeda, K.; Koike, T. Detection of noncarboxymethyllysine and carboxymethyllysine advanced glycation end products (AGE) in serum of diabetic patients. Mol. Med. 1999, 5, 393–405. [Google Scholar] [PubMed]
- Takeuchi, M.; Makita, Z.; Bucala, R.; Suzuki, T.; Koike, T.; Kameda, Y. Immunological evidence that non-carboxymethyllysine advanced glycation end-products are produced from short chain sugars and dicarbonyl compounds in vivo. Mol. Med. 2000, 6, 114–125. [Google Scholar] [PubMed]
- Takeuchi, M.; Yanase, Y.; Matsuura, N.; Yamagishi, S.; Kameda, Y.; Bucala, R.; Makita, Z. Immunological detection of a novel advanced glycation end-product. Mol. Med. 2001, 7, 783–791. [Google Scholar] [PubMed]
- Takeuchi, M.; Iwaki, M.; Takino, J.; Shirai, H.; Kawakami, M.; Bucala, R.; Yamagishi, S. Immunological detection of fructose-derived advanced glycation end-products. Lab. Investig. 2010, 90, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Koschinsky, T.; He, C.; Mitsuhashi, T.; Bucala, R.; Liu, C.; Buenting, C.; Heitmann, K.; Vlassara, H. Orally absorbed reactive glycation products (glycotoxins): An environmental risk factor in diabetic nephropathy. Proc. Natl. Acad. Sci. USA 1997, 94, 6474–6479. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T. Pathologic role of dietary advanced glycation end products in cardiometabolic disorders, and therapeutic intervention. Nutrition 2016, 32, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Uribarri, J. Dietary advanced glycation end products and their potential role in cardiometabolic disease in children. Horm. Res. Paediatr. 2016, 85, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Linnane, A.W.; Kios, M.; Vitetta, L. Healthy aging: Regulation of the metabolome by cellular redox modulation and prooxidant signaling systems: The essential roles of superoxide anion and hydrogen peroxide. Biogerontology 2007, 8, 445–467. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| (g per Bottle/Can/Glass) | |||||
|---|---|---|---|---|---|
| Sugar Content | ≥25 | 12.5–24.9 | <12.5 | ||
| (Average) | (Min–Max) | ||||
| Alcoholic beverages (135) | 18.5 | ||||
| Effervescent alcoholic beverages (21) | 16.4 | (5.6–23.8) | 14 | 7 | |
| Brewed alcoholic beverages (27) | 12.2 | (6.6–24.1) | 11 | 16 | |
| Distilled alcoholic beverages (16) | 7.0 | (2.9–12.3) | 16 | ||
| Mixed liquor (71) | 24.1 | (5.0–43.8) | 32 | 34 | 5 |
| (Number of alcoholic beverages) | (32) | (59) | (44) | ||
| (U per Bottle/Can/Glass) | |||||
|---|---|---|---|---|---|
| Glu-AGE Content | ≥50,000 | 20,000–49,999 | <20,000 | ||
| (Average) | (Min–Max) | ||||
| Alcoholic beverages (135): | 17,750 | ||||
| Effervescent alcoholic beverages (21) | 1010 | (0–9210) | 21 | ||
| Brewed alcoholic beverages (27) | 2600 | (0–23,290) | 1 | 26 | |
| Distilled alcoholic beverages (16) | 9430 | (0–26,210) | 4 | 12 | |
| Mixed liquor (71) | 30,340 | (0–64,710) | 6 | 45 | 20 |
| (Number of alcoholic beverages) | (6) | (50) | (79) | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeuchi, M.; Takino, J.-i.; Sakasai-Sakai, A.; Takata, T.; Tsutsumi, M. Toxic AGE (TAGE) Theory for the Pathophysiology of the Onset/Progression of NAFLD and ALD. Nutrients 2017, 9, 634. https://doi.org/10.3390/nu9060634
Takeuchi M, Takino J-i, Sakasai-Sakai A, Takata T, Tsutsumi M. Toxic AGE (TAGE) Theory for the Pathophysiology of the Onset/Progression of NAFLD and ALD. Nutrients. 2017; 9(6):634. https://doi.org/10.3390/nu9060634
Chicago/Turabian StyleTakeuchi, Masayoshi, Jun-ichi Takino, Akiko Sakasai-Sakai, Takanobu Takata, and Mikihiro Tsutsumi. 2017. "Toxic AGE (TAGE) Theory for the Pathophysiology of the Onset/Progression of NAFLD and ALD" Nutrients 9, no. 6: 634. https://doi.org/10.3390/nu9060634
APA StyleTakeuchi, M., Takino, J. -i., Sakasai-Sakai, A., Takata, T., & Tsutsumi, M. (2017). Toxic AGE (TAGE) Theory for the Pathophysiology of the Onset/Progression of NAFLD and ALD. Nutrients, 9(6), 634. https://doi.org/10.3390/nu9060634