LXRα Regulates Hepatic ChREBPα Activity and Lipogenesis upon Glucose, but Not Fructose Feeding in Mice

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals and Treatment
2.3. Blood Chemistries
2.4. Metabolomics
2.5. RNA Extraction, cDNA Synthesis and Real-Time Quantitative PCR (RT-qPCR)
2.6. Liver Extracts and Immunoblot Analysis
2.7. Chromatin Immunoprecipitation (ChIP)
2.8. Wheat Germ Agglutinin (WGA) Pulldown
2.9. UDP-GlcNAc Measurements
2.10. Statistical Analysis
3. Results
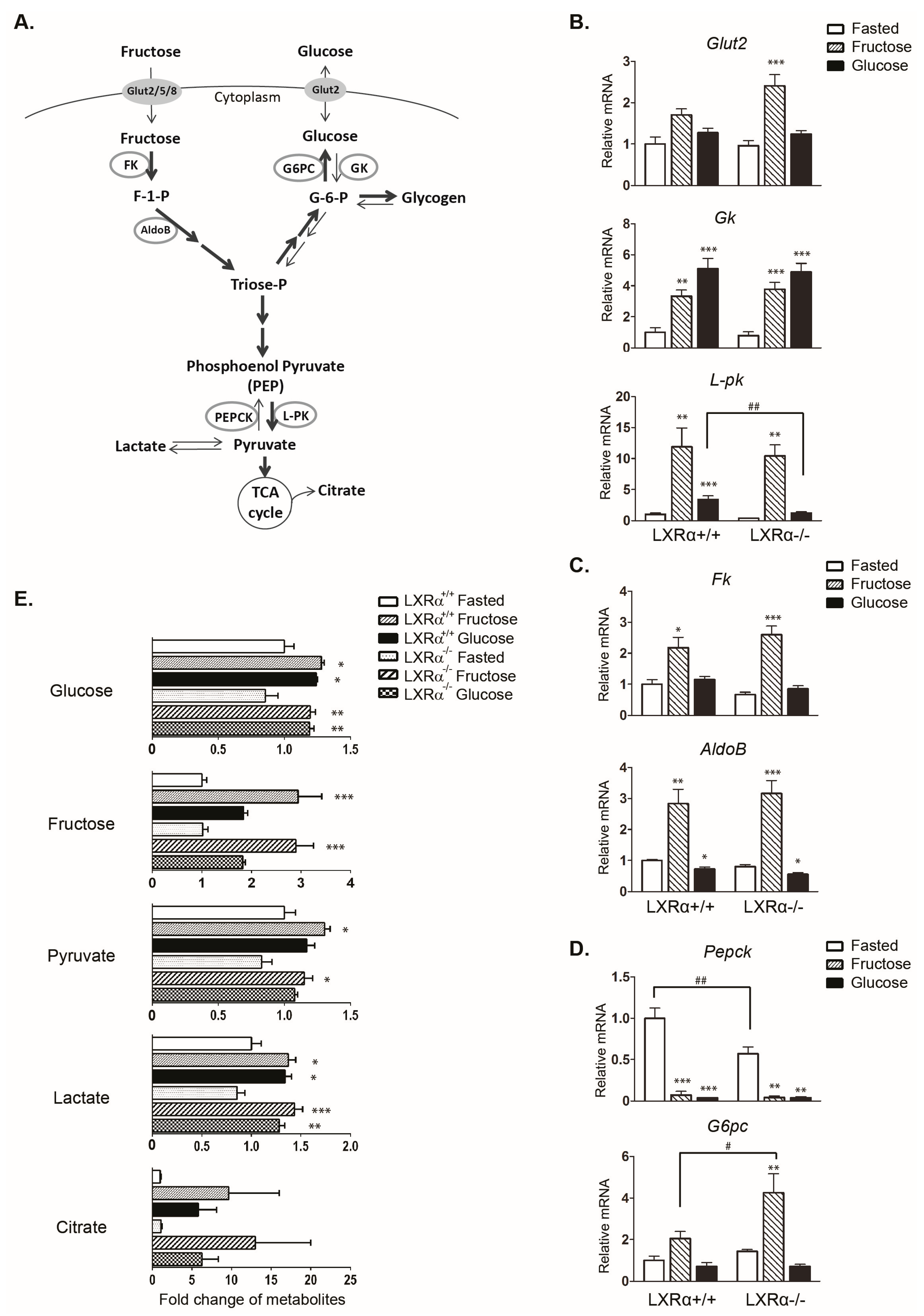
3.1. Regulation of Genes Involved in Carbohydrate Metabolism by Dietary Fructose and Glucose-Role of LXRα
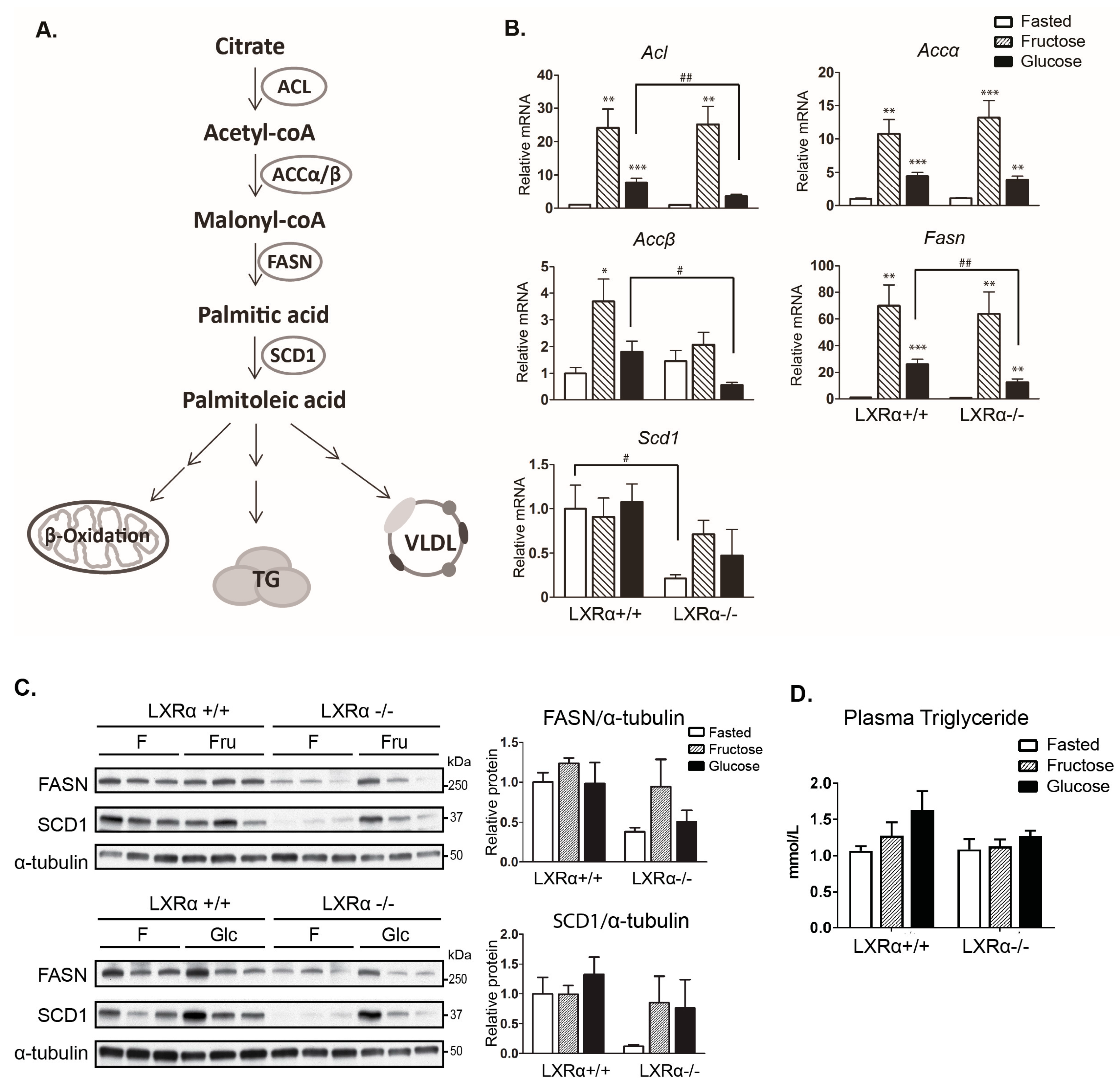
3.2. Expression of Hepatic de novo Lipogenic Genes Mediated by Dietary Glucose Is Reduced in LXRα−/− Compared to Wild Type Mice
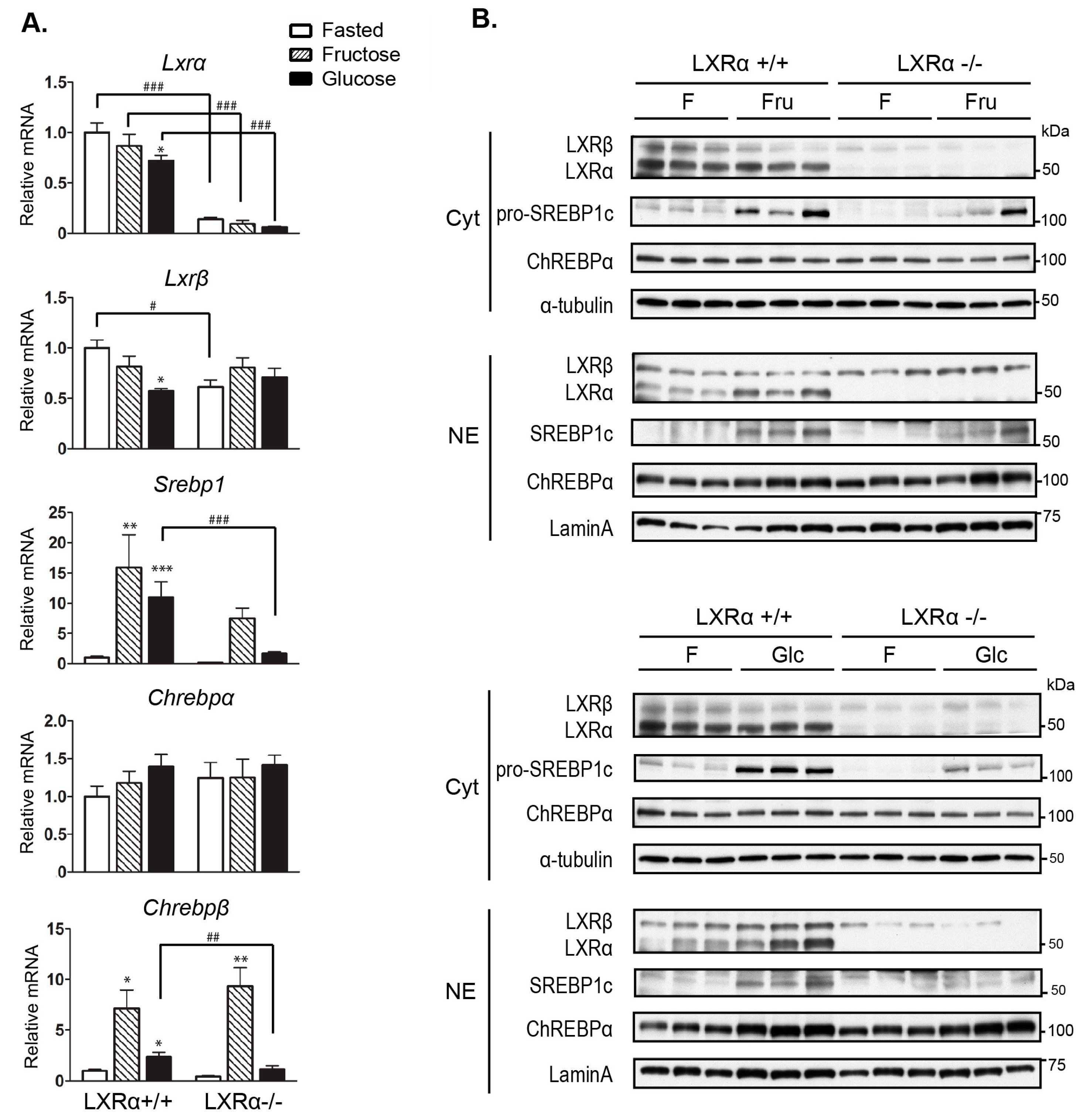
3.3. Regulation of Srebp-1 and Chrebpβ Expression Mediated by Dietary Glucose Is Dependent of LXRα
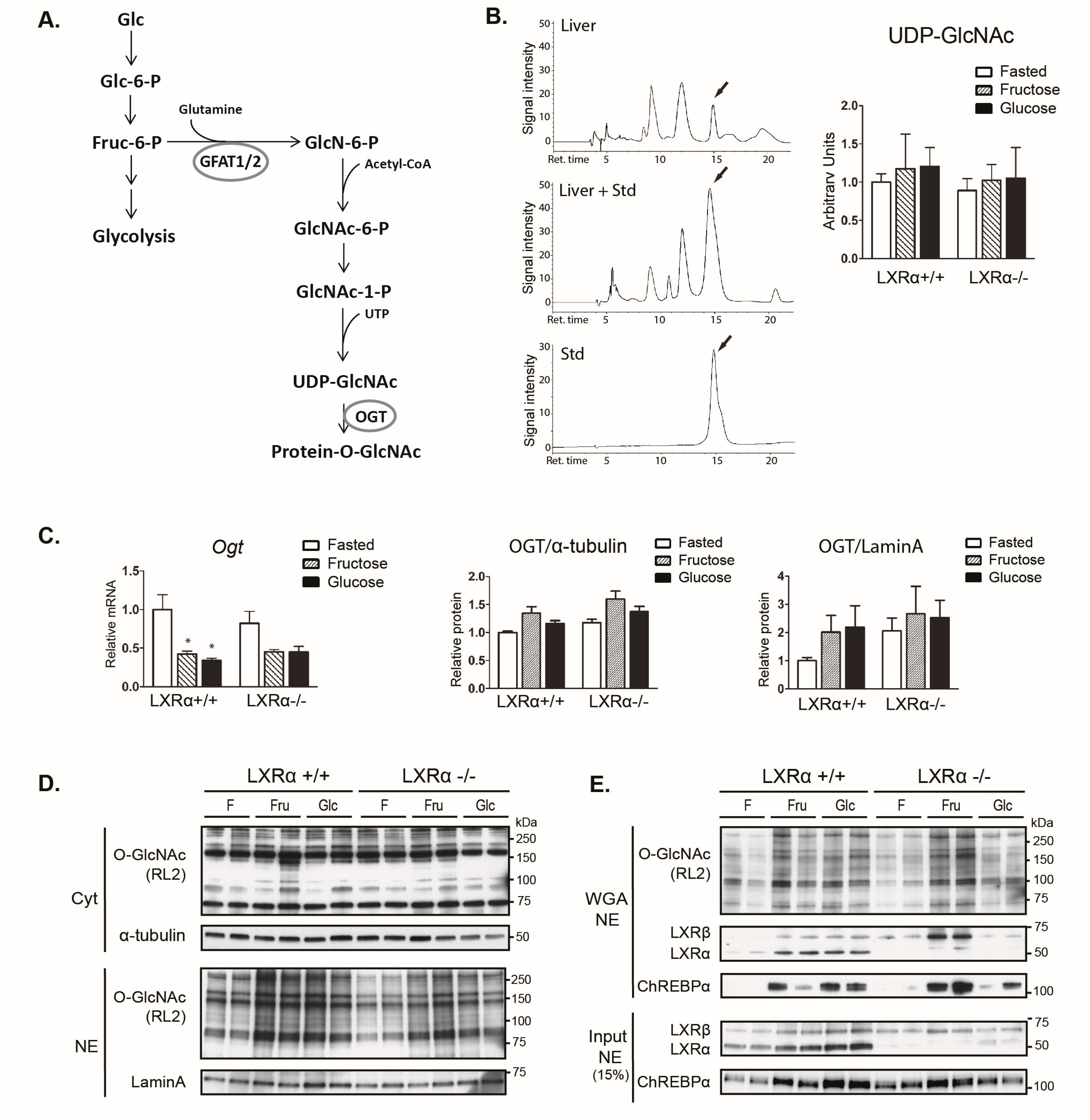
3.4. Dietary Fructose Induces Nuclear O-GlcNAc Signaling
4. Discussion and Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bantle, J.P.; Raatz, S.K.; Thomas, W.; Georgopoulos, A. Effects of dietary fructose on plasma lipids in healthy subjects. Am. J. Clin. Nutr. 2000, 72, 1128–1134. [Google Scholar] [PubMed]
- Parks, E.J.; Hellerstein, M.K. Carbohydrate-induced hypertriacylglycerolemia: Historical perspective and review of biological mechanisms. Am. J. Clin. Nutr. 2000, 71, 412–433. [Google Scholar] [PubMed]
- Faeh, D.; Minehira, K.; Schwarz, J.M.; Periasamy, R.; Park, S.; Tappy, L. Effect of fructose overfeeding and fish oil administration on hepatic de novo lipogenesis and insulin sensitivity in healthy men. Diabetes 2005, 54, 1907–1913. [Google Scholar] [CrossRef] [PubMed]
- Caton, P.W.; Nayuni, N.K.; Khan, N.Q.; Wood, E.G.; Corder, R. Fructose induces gluconeogenesis and lipogenesis through a SIRT1-dependent mechanism. J. Endocrinol. 2011, 208, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Strable, M.S.; Ntambi, J.M. Genetic control of de novo lipogenesis: Role in diet-induced obesity. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Grønning-Wang, L.M.; Bindesbøll, C.; Nebb, H.I. The Role of Liver X Receptor in Hepatic de novo Lipogenesis and Cross-Talk with Insulin and Glucose Signaling; INTECH Open Access Publisher: Rijeka, Croatia, 2013. [Google Scholar]
- Repa, J.J.; Mangelsdorf, D.J. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu. Rev. Cell Dev. Biol. 2000, 16, 459–481. [Google Scholar] [CrossRef] [PubMed]
- Filhoulaud, G.; Guilmeau, S.; Dentin, R.; Girard, J.; Postic, C. Novel insights into ChREBP regulation and function. Trends Endocrinol. Metab. 2013, 24, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Laffitte, B.A.; Chao, L.C.; Li, J.; Walczak, R.; Hummasti, S.; Joseph, S.B.; Castrillo, A.; Wilpitz, D.C.; Mangelsdorf, D.J.; Collins, J.L.; et al. Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue. Proc. Natl. Acad. Sci. USA 2003, 100, 5419–5424. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Liang, Y.; Broderick, C.L.; Oldham, B.A.; Beyer, T.P.; Schmidt, R.J.; Zhang, Y.; Stayrook, K.R.; Suen, C.; Otto, K.A.; et al. Antidiabetic action of a liver x receptor agonist mediated by inhibition of hepatic gluconeogenesis. J. Boil. Chem. 2003, 278, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Grempler, R.; Gunther, S.; Steffensen, K.R.; Nilsson, M.; Barthel, A.; Schmoll, D.; Walther, R. Evidence for an indirect transcriptional regulation of glucose-6-phosphatase gene expression by liver X receptors. Biochem. Biophys. Res. Commun. 2005, 338, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Anthonisen, E.H.; Berven, L.; Holm, S.; Nygard, M.; Nebb, H.I.; Gronning-Wang, L.M. Nuclear Receptor Liver X Receptor Is O-GlcNAc-modified in Response to Glucose. J. Biol. Chem. 2010, 285, 1607–1615. [Google Scholar] [CrossRef] [PubMed]
- Guinez, C.; Filhoulaud, G.; Rayah-Benhamed, F.; Marmier, S.; Dubuquoy, C.; Dentin, R.; Moldes, M.; Burnol, A.F.; Yang, X.; Lefebvre, T.; et al. O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 2011, 60, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.R.; Hanover, J.A. A little sugar goes a long way: The cell biology of O-GlcNAc. J. Cell Biol. 2015, 208, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.W.; Slawson, C.; Ramirez-Correa, G.; Lagerlof, O. Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 2011, 80, 825–858. [Google Scholar] [CrossRef] [PubMed]
- Gambetta, M.C.; Muller, J. A critical perspective of the diverse roles of O-GlcNAc transferase in chromatin. Chromosoma 2015, 124, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Rumberger, J.M.; Wu, T.; Hering, M.A.; Marshall, S. Role of hexosamine biosynthesis in glucose-mediated up-regulation of lipogenic enzyme mRNA levels: Effects of glucose, glutamine, and glucosamine on glycerophosphate dehydrogenase, fatty acid synthase, and acetyl-CoA carboxylase mRNA levels. J. Biol. Chem. 2003, 278, 28547–28552. [Google Scholar] [CrossRef] [PubMed]
- Bindesbøll, C.; Grønning-Wang, L.M. Liver X receptors connect nuclear O-GlcNAc signaling to hepatic glucose utilization and lipogenesis. Recept. Clin. Investig. 2015. [Google Scholar] [CrossRef]
- Bindesboll, C.; Fan, Q.; Norgaard, R.C.; MacPherson, L.; Ruan, H.B.; Wu, J.; Pedersen, T.A.; Steffensen, K.R.; Yang, X.; Matthews, J.; et al. Liver X receptor regulates hepatic nuclear O-GlcNAc signaling and carbohydrate responsive element-binding protein activity. J. Lipid Res. 2015, 56, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schon, M.R.; Abumrad, N.A.; Bluher, M.; Klein, S.; Kahn, B.B. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012, 484, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Stamatikos, A.D.; da Silva, R.P.; Lewis, J.T.; Douglas, D.N.; Kneteman, N.M.; Jacobs, R.L.; Paton, C.M. Tissue Specific Effects of Dietary Carbohydrates and Obesity on ChREBPα and ChREBPβ Expression. Lipids 2016, 51, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Kumar, A.; Katz, L.S.; Li, L.; Paulynice, M.; Herman, M.A.; Scott, D.K. Induction of the ChREBPβ Isoform Is Essential for Glucose-Stimulated β-Cell Proliferation. Diabetes 2015, 64, 4158–4170. [Google Scholar] [CrossRef] [PubMed]
- Erion, D.M.; Popov, V.; Hsiao, J.J.; Vatner, D.; Mitchell, K.; Yonemitsu, S.; Nagai, Y.; Kahn, M.; Gillum, M. P.; Dong, J.; et al. The role of the carbohydrate response element-binding protein in male fructose-fed rats. Endocrinology 2013, 154, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Krawczyk, S.A.; Doridot, L.; Fowler, A.J.; Wang, J.X.; Trauger, S.A.; Noh, H.L.; Kang, H.J.; Meissen, J.K.; Blatnik, M.; et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J. Clin. Investig. 2016, 126, 4372–4386. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Boekschoten, M.V.; Baltzersen, M.A.; Kersten, S.; Xu, X.; Andersen, H.; Rustan, A.C.; Thoresen, G.H. LXRβ is the dominant LXR subtype in skeletal muscle regulating lipogenesis and cholesterol efflux. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E602–E613. [Google Scholar] [CrossRef] [PubMed]
- Rull, A.; Geeraert, B.; Aragones, G.; Beltran-Debon, R.; Rodriguez-Gallego, E.; Garcia-Heredia, A.; Pedro-Botet, J.; Joven, J.; Holvoet, P.; Camps, J. Rosiglitazone and fenofibrate exacerbate liver steatosis in a mouse model of obesity and hyperlipidemia. A transcriptomic and metabolomic study. J. Proteome Res. 2014, 13, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.P.; Akimoto, Y.; Hart, G.W. Identification and cloning of a novel family of coiled-coil domain proteins that interact with O-GlcNAc transferase. J. Biol. Chem. 2003, 278, 5399–5409. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.K.; Chong, H.K.; Infante, A.M.; Im, S.S.; Xie, X.; Osborne, T.F. Genome-wide analysis of SREBP-1 binding in mouse liver chromatin reveals a preference for promoter proximal binding to a new motif. Proc. Natl. Acad. Sci. USA 2009, 106, 13765–13769. [Google Scholar] [CrossRef] [PubMed]
- Itkonen, H.M.; Engedal, N.; Babaie, E.; Luhr, M.; Guldvik, I.J.; Minner, S.; Hohloch, J.; Tsourlakis, M.C.; Schlomm, T.; Mills, I.G. UAP1 is overexpressed in prostate cancer and is protective against inhibitors of N-linked glycosylation. Oncogene 2015, 34, 3744–3750. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Kitazume, S.; Angata, T.; Fujinawa, R.; Ohtsubo, K.; Miyoshi, E.; Taniguchi, N. Simultaneous determination of nucleotide sugars with ion-pair reversed-phase HPLC. Glycobiology 2010, 20, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.Y.; Wallig, M.A.; Chung, B.H.; Nara, T.Y.; Cho, B.H.; Nakamura, M.T. Dietary fructose induces a wide range of genes with distinct shift in carbohydrate and lipid metabolism in fed and fasted rat liver. Biochim. Biophys. Acta 2008, 1782, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaka, T.; Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Ohashi, K.; Tomita, S.; Sekiya, M.; et al. Insulin-independent induction of sterol regulatory element-binding protein-1c expression in the livers of streptozotocin-treated mice. Diabetes 2004, 53, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Ishimoto, T.; Li, N.; Cicerchi, C.; Orlicky, D.J.; Ruzycki, P.; Rivard, C.; Inaba, S.; Roncal-Jimenez, C.A.; Bales, E.S.; et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat. Commun. 2013, 4, 2434. [Google Scholar] [CrossRef] [PubMed]
- Tounian, P.; Schneiter, P.; Henry, S.; Jequier, E.; Tappy, L. Effects of infused fructose on endogenous glucose production, gluconeogenesis, and glycogen metabolism. Am. J. Physiol. 1994, 267, E710–E717. [Google Scholar] [CrossRef]
- Rippe, J.M.; Angelopoulos, T.J. Sucrose, high-fructose corn syrup, and fructose, their metabolism and potential health effects: What do we really know? Adv. Nutr. Int. Rev. J. 2013, 4, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Janevski, M.; Ratnayake, S.; Siljanovski, S.; McGlynn, M.A.; Cameron-Smith, D.; Lewandowski, P. Fructose containing sugars modulate mRNA of lipogenic genes ACC and FAS and protein levels of transcription factors ChREBP and SREBP1c with no effect on body weight or liver fat. Food Funct. 2012, 3, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.Y.; Miyashita, M.; Cho, B.H.; Nakamura, M.T. Replacing dietary glucose with fructose increases ChREBP activity and SREBP-1 protein in rat liver nucleus. Biochem. Biophys. Res. Commun. 2009, 390, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Chu, K.; Miyazaki, M.; Man, W.C.; Ntambi, J.M. Stearoyl-coenzyme A desaturase 1 deficiency protects against hypertriglyceridemia and increases plasma high-density lipoprotein cholesterol induced by liver X receptor activation. Mol. Cell. Biol. 2006, 26, 6786–6798. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Riesland, L.; Paterson, A.J.; Kudlow, J.E. Phosphorylation of mouse glutamine-fructose-6-phosphate amidotransferase 2 (GFAT2) by cAMP-dependent protein kinase increases the enzyme activity. J. Biol. Chem. 2004, 279, 29988–29993. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.P.; Geisler, T.S.; Chambers, J.H.; McClain, D.A. Up-regulation of O-GlcNAc transferase with glucose deprivation in HepG2 cells is mediated by decreased hexosamine pathway flux. J. Boil. Chem. 2009, 284, 3425–3432. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Iizuka, K.; Miller, B.C.; Uyeda, K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 15597–15602. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.C.; Coate, K.C.; Winnick, J.J.; An, Z.; Cherrington, A.D. Regulation of hepatic glucose uptake and storage in vivo. Adv. Nutr. (Bethesda, Md.) 2012, 3, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Z.; Empie, M.W. Fructose metabolism in humans—What isotopic tracer studies tell us. Nutr. Metab. 2012, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Parks, E.J.; Skokan, L.E.; Timlin, M.T.; Dingfelder, C.S. Dietary sugars stimulate fatty acid synthesis in adults. J. Nutr. 2008, 138, 1039–1046. [Google Scholar] [PubMed]
- Moore, J.B.; Gunn, P.J.; Fielding, B.A. The role of dietary sugars and de novo lipogenesis in non-alcoholic fatty liver disease. Nutrients 2014, 6, 5679–5703. [Google Scholar] [CrossRef] [PubMed]
- Quinet, E.M.; Savio, D.A.; Halpern, A.R.; Chen, L.; Schuster, G.U.; Gustafsson, J.A.; et al. Liver X receptor (LXR)-β regulation in LXRα-deficient mice: Implications for therapeutic targeting. Mol. Pharmacol. 2006, 70, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, S.; Blander, G.; Tse, J.G.; Krieger, M.; Guarente, L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol. Cell 2007, 28, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Bricambert, J.; Miranda, J.; Benhamed, F.; Girard, J.; Postic, C.; Dentin, R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J. Clin. Investig. 2010, 120, 4316–4331. [Google Scholar] [CrossRef] [PubMed]
- Allison, D.F.; Wamsley, J.J.; Kumar, M.; Li, D.; Gray, L.G.; Hart, G.W.; Jones, D.R.; Mayo, M.W. Modification of RelA by O-linked N-acetylglucosamine links glucose metabolism to NF-κB acetylation and transcription. Proc. Natl. Acad. Sci. USA 2012, 109, 16888–16893. [Google Scholar] [CrossRef] [PubMed]
- Oosterveer, M.H.; Schoonjans, K. Hepatic glucose sensing and integrative pathways in the liver. Cell. Mol. Life Sci. CMLS 2014, 71, 1453–1467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Yin, R.; Yang, X. O-GlcNAc: A Bittersweet Switch in Liver. Front. Endocrinol. 2014, 5, 221. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Treatment | Body Weight (g) | Food Intake (g) | Plasma Insulin (μg/dL) |
|---|---|---|---|---|
| LXRα+/+ | Fasted | 30.8 ± 2.0 | 0.7 ± 0.1 | |
| Refed-Fructose | 28.4 ± 1.7 | 2.4 ± 0.4 | 2.0 ± 0.7 | |
| Refed-Glucose | 29.6 ± 2.3 | 3.3 ± 0.2 | 7.0 ± 1.0 *** | |
| LXRα−/− | Fasted | 32.7 ± 1.5 | 0.7 ± 0.1 | |
| Refed-Fructose | 31.1 ± 2.9 | 2.8 ± 0.2 | 5.0 ± 1.5 * | |
| Refed-Glucose | 32.1 ± 3.5 | 2.7 ± 0.4 | 7.1 ± 0.8 *** |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Q.; Nørgaard, R.C.; Bindesbøll, C.; Lucas, C.; Dalen, K.T.; Babaie, E.; Itkonen, H.M.; Matthews, J.; Nebb, H.I.; Grønning-Wang, L.M. LXRα Regulates Hepatic ChREBPα Activity and Lipogenesis upon Glucose, but Not Fructose Feeding in Mice. Nutrients 2017, 9, 678. https://doi.org/10.3390/nu9070678
Fan Q, Nørgaard RC, Bindesbøll C, Lucas C, Dalen KT, Babaie E, Itkonen HM, Matthews J, Nebb HI, Grønning-Wang LM. LXRα Regulates Hepatic ChREBPα Activity and Lipogenesis upon Glucose, but Not Fructose Feeding in Mice. Nutrients. 2017; 9(7):678. https://doi.org/10.3390/nu9070678
Chicago/Turabian StyleFan, Qiong, Rikke C. Nørgaard, Christian Bindesbøll, Christin Lucas, Knut Tomas Dalen, Eshrat Babaie, Harri M. Itkonen, Jason Matthews, Hilde I. Nebb, and Line M. Grønning-Wang. 2017. "LXRα Regulates Hepatic ChREBPα Activity and Lipogenesis upon Glucose, but Not Fructose Feeding in Mice" Nutrients 9, no. 7: 678. https://doi.org/10.3390/nu9070678
APA StyleFan, Q., Nørgaard, R. C., Bindesbøll, C., Lucas, C., Dalen, K. T., Babaie, E., Itkonen, H. M., Matthews, J., Nebb, H. I., & Grønning-Wang, L. M. (2017). LXRα Regulates Hepatic ChREBPα Activity and Lipogenesis upon Glucose, but Not Fructose Feeding in Mice. Nutrients, 9(7), 678. https://doi.org/10.3390/nu9070678