Role of Mitochondria and Endoplasmic Reticulum in Taurine-Deficiency-Mediated Apoptosis





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Model of Taurine Deficiency (TauTKO)
2.2. Measurement of Mitochondrial Taurine Content
2.3. Assay of Respiratory Chain Complexes
2.4. Quantitative Real Time PCR Method of Measuring Levels of ND6 mRNA
2.5. Assay of Aconitase Activity
2.6. Determination of Glutathione Redox State
2.7. Measurement of Protein Carbonylation
2.8. Determination of Protein Content via Western Blot Analysis
2.9. Evaluation of Mitochondrial Oxidative Stress
2.10. Statistical Analyses
3. Results
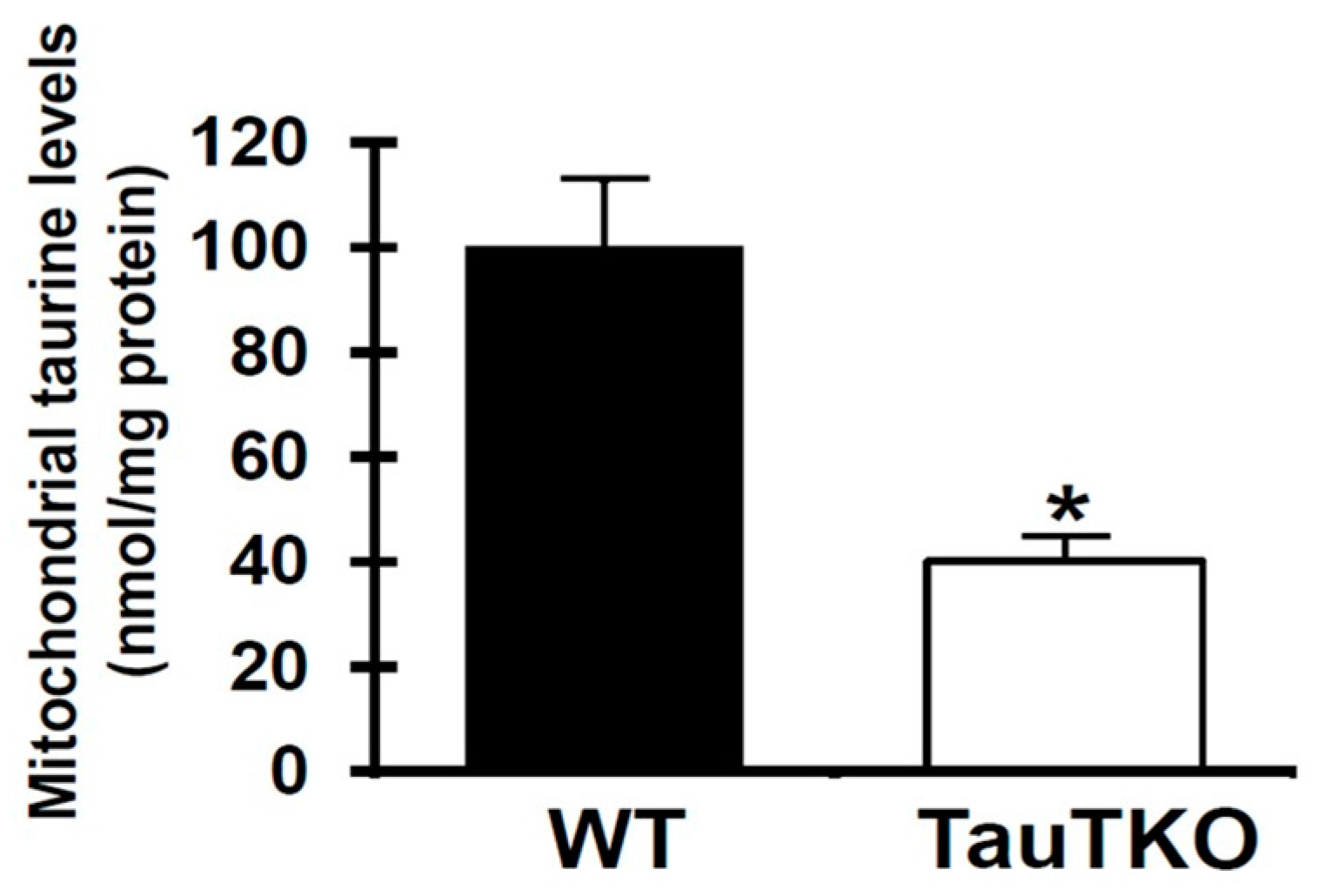
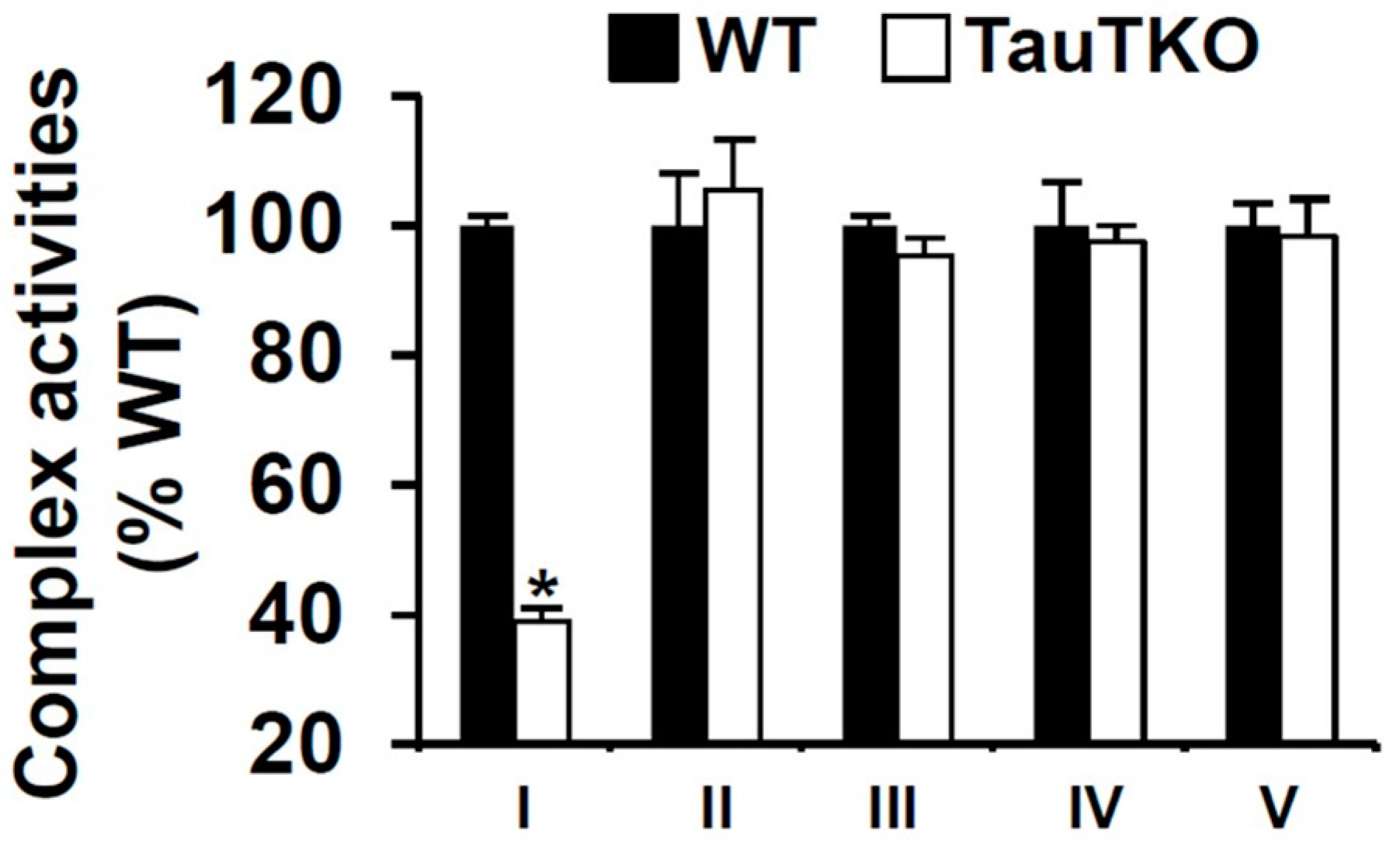
3.1. Effect of Taurine on Mitochondrial Function
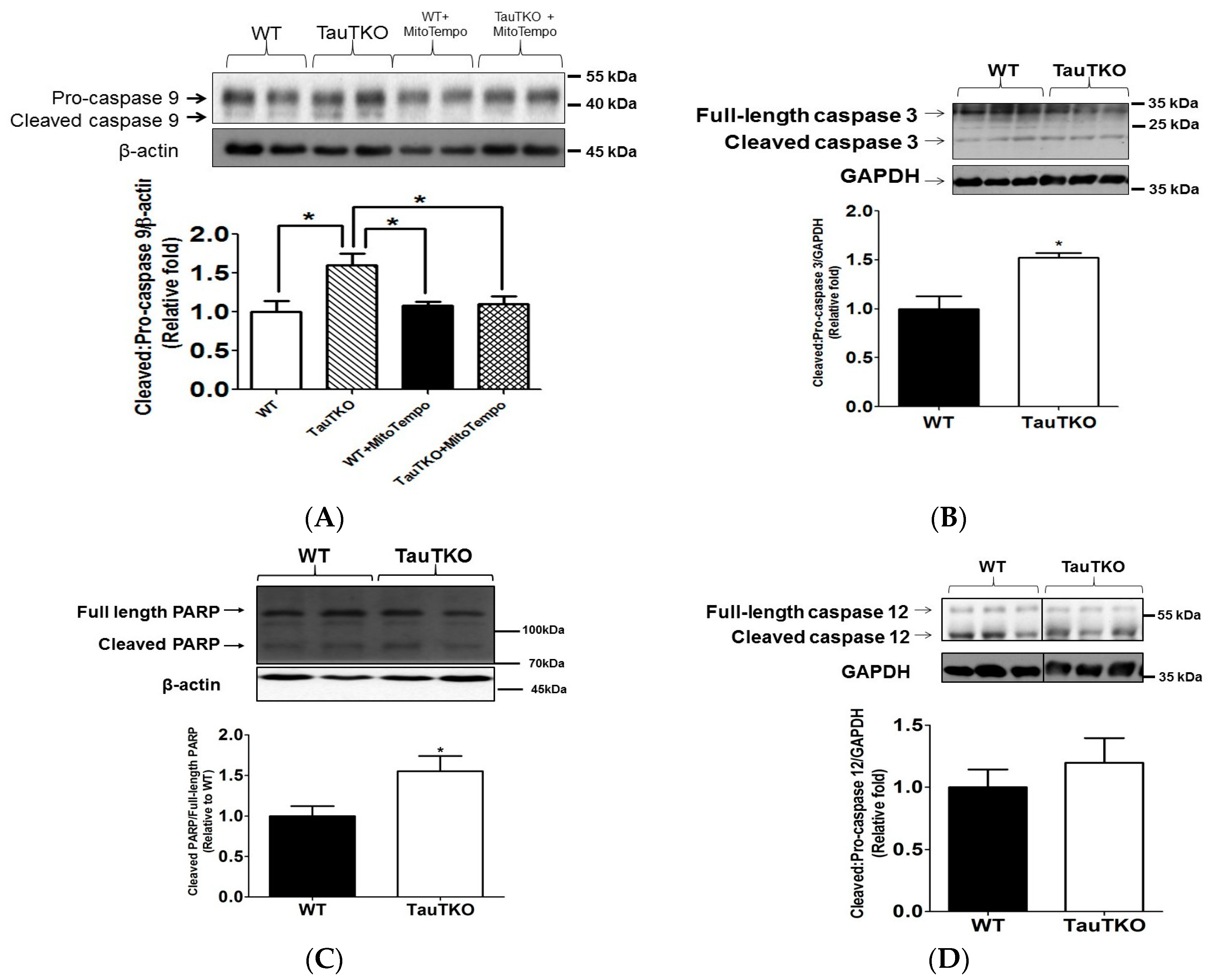
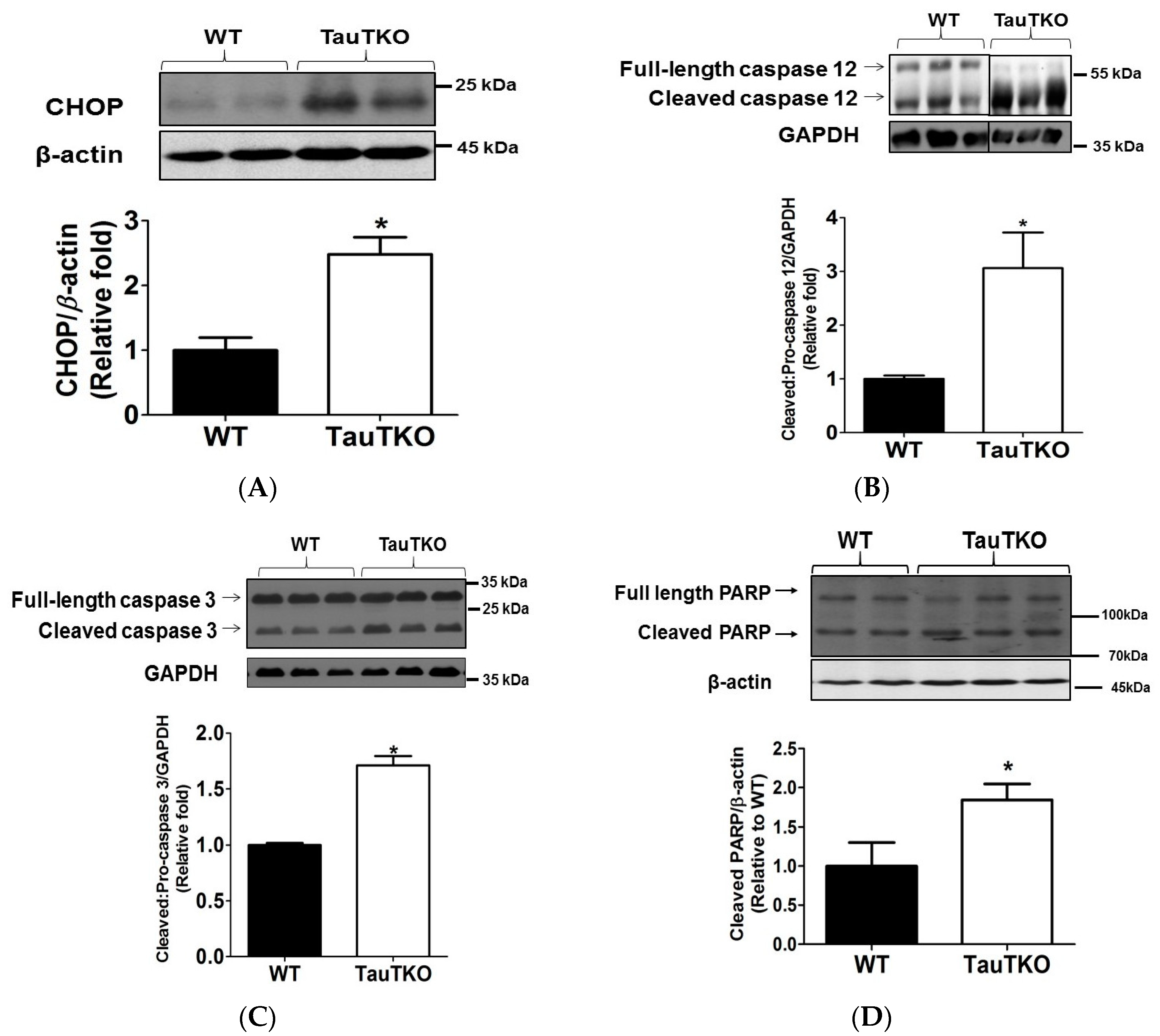
3.2. Taurine Depletion Induces Cell Death—An Effect Mediated by Oxidative Stress
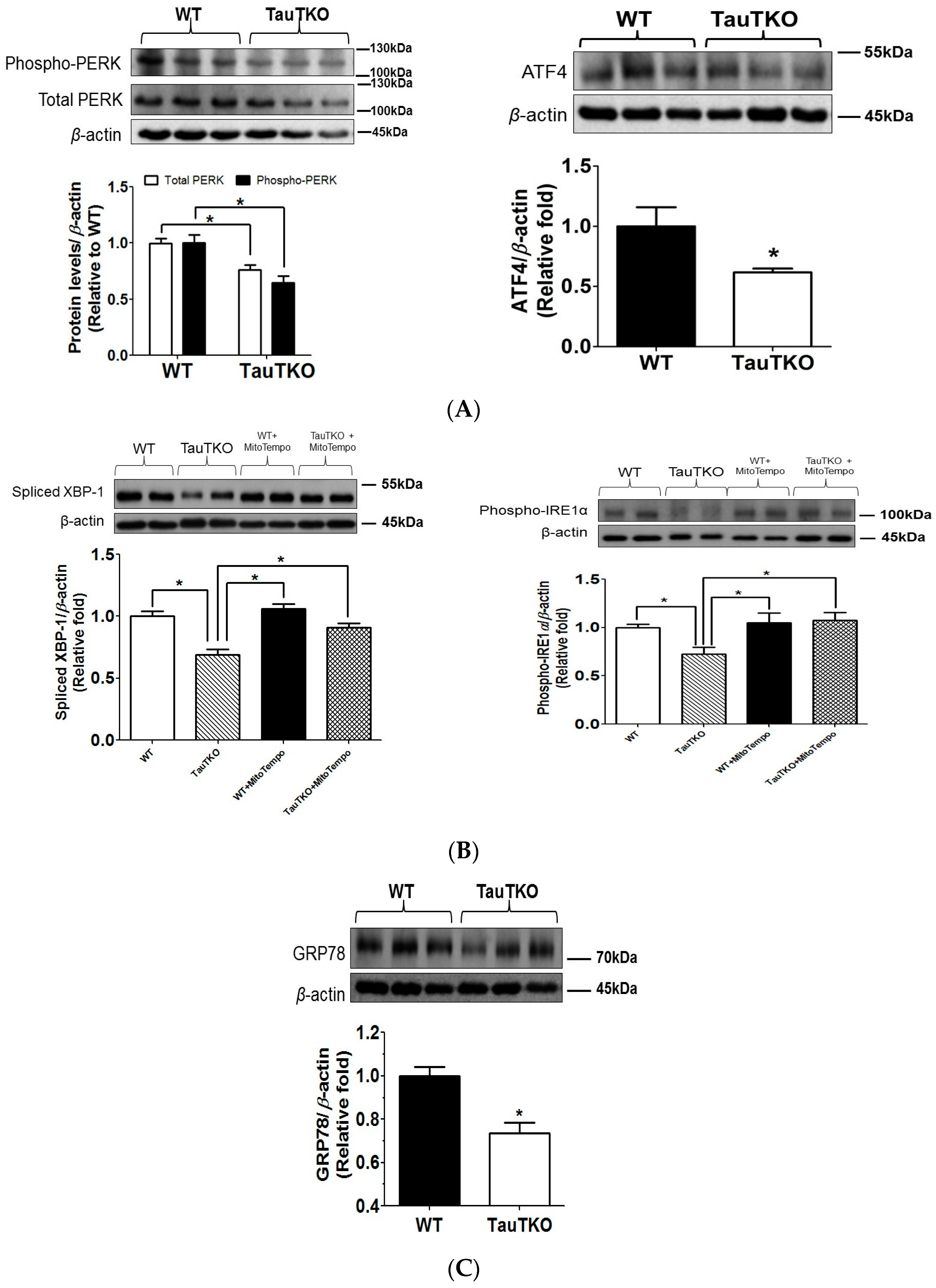
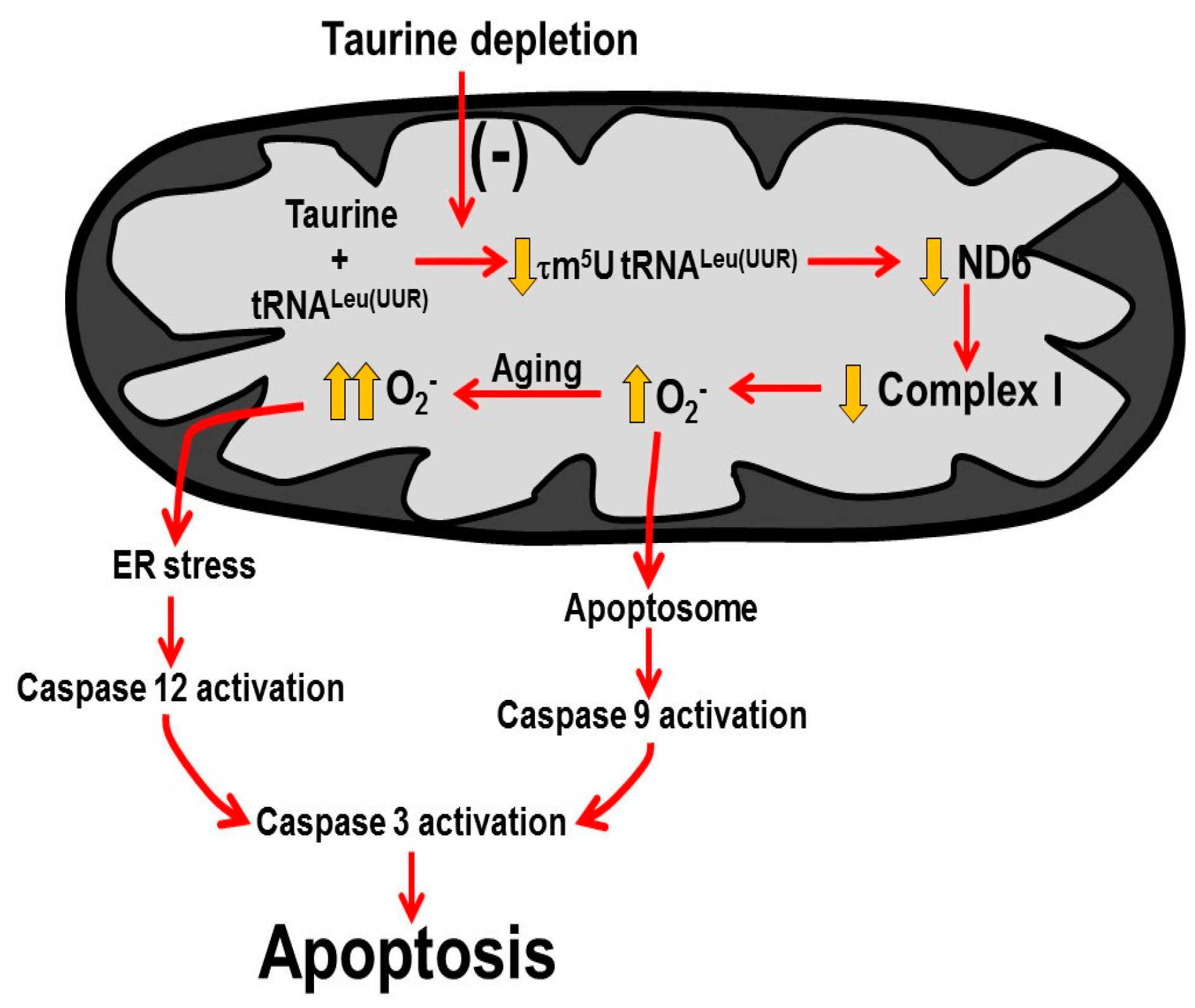
3.3. Potential Crosstalk between Mitochondria and ER
4. Discussion
4.1. Mitochondrial Actions of Taurine
4.2. Role of ER Stress in TauTKO Hearts
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Gaull, G.E. Taurine as a conditionally essential nutrient in man. J. Am. Coll. Nutr. 1986, 5, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Yamori, Y.; Taguchi, T.; Hamada, A.; Kunimasa, K.; Mori, H.; Mori, M. Taurine in health and diseases: Consistent evidence from experimental and epidemiological studies. J. Biomed. Sci. 2010, 17, S6. [Google Scholar] [CrossRef] [PubMed]
- Hayes, K.C.; Carey, R.E.; Schmidt, S.Y. Retinal degeneration associated with taurine deficiency in the cat. Science 1975, 188, 949–951. [Google Scholar] [CrossRef] [PubMed]
- Pion, P.D.; Kittleson, M.D.; Rogers, Q.R.; Morris, J.G. Myocardial failure in cats associated with low plasma taurine: A reversible cardiomyopathy. Science 1987, 237, 764–768. [Google Scholar] [CrossRef] [PubMed]
- Hayes, K.C. Taurine requirement in primates. Nutr. Rev. 1985, 43, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Kimura, Y.; Uozumi, Y.; Takai, M.; Muraoka, S.; Matsuda, T.; Uyeki, K.; Yoshiyama, M.; Ikawa, M.; Okabe, M.; et al. Taurine depletion caused by knocking out the taurine transporter gene leads to cardiomyopathy with cardiac atrophy. J. Mol. Cell. Cardiol. 2008, 44, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Jong, C.J.; Takahashi, K.; Schaffer, S.W. Role of ROS production and turnover in the antioxidant activity of taurine. Adv. Exp. Med. Biol. 2015, 803, 581–596. [Google Scholar] [PubMed]
- Marcinkiewicz, J.; Kontny, E. Taurine and inflammatory diseases. Amino Acids 2014, 46, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Shetewy, A.; Shimada-Takaura, K.; Warner, D.; Jong, C.J.; Mehdi, A.B.; Alexeyev, M.; Takahashi, K.; Schaffer, S.W. Mitochondrial defects associated with β-alanine toxicity: Relevance to hyper-beta-alaninemia. Mol. Cell. Biochem. 2016, 416, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Ricci, C.; Pastukh, V.; Leonard, J.; Turrens, J.; Wilson, G.; Schaffer, D.; Schaffer, S.W. Mitochondrial DNA damage triggers mitochondrial superoxide generation and apoptosis. Am. J. Physiol. 2008, 294, C413–C422. [Google Scholar] [CrossRef] [PubMed]
- Ramila, K.C.; Jong, C.J.; Pastukh, V.; Ito, T.; Azuma, J.; Schaffer, S.W. Role of protein phosphorylation in excitation-contraction coupling in taurine deficient hearts. Am. J. Physiol. 2015, 308, H232–H239. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Grishko, V.; Pastukh, V.; Solodushko, V.; Gillespie, M.; Azuma, J.; Schaffer, S. Apoptotic cascade initiated by angiotensin II in neonatal cardiomyocytes: Role of DNA damage. Am. J. Physiol. 2003, 285, H2364–H2372. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, J.E.; Kocsis, J.J. Taurine mobilizing effects of beta alanine and other inhibitors of taurine transport. Life Sci. 1981, 28, 2727–2736. [Google Scholar] [CrossRef]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of coomplex III. J. Biol. Chem. 2003, 278, 36927–36931. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.Y.; Zhang, X.L.; Wu, T.F.; Liu, Y.P.; Wang, Q.; Zhang, Y.; Song, J.Q.; Wang, Y.J.; Yang, Y.L. Analysis of the mitochondrial complexes I–V enzyme activities of peripheral leukocytes in oxidative phosphorylation disorder. J. Child Neurol. 2011, 26, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, S.W.; Seyed-Mozaffari, M.; Cutcliff, C.R.; Wilson, G.L. Postreceptor myocardial defect in a rat model of non-insulin-dependent diabetes mellitus. Diabetes 1986, 55, 593–597. [Google Scholar] [CrossRef]
- Suzuki, T.; Suzuki, T.; Wada, T.; Saigo, K.; Watanabe, K. Taurine as a constituent of mitochondrial tRNAs: New insights into the functions of taurine and human mitochondrial diseases. EMBO J. 2002, 21, 6581–6589. [Google Scholar] [CrossRef] [PubMed]
- Kurata, S.; Ohtsuki, T.; Wada, T.; Kirino, Y.; Takai, K.; Sigo, K.; Watanabe, K.; Suzuki, T. Decoding property of C5 uridine modification at the wobble position of tRNA anticodon. Nucleic Acids Res. 2003, 3, 245–246. [Google Scholar] [CrossRef]
- Schaffer, S.W.; Jong, C.J.; Ito, T.; Azuma, J. Role of taurine in the pathogenesis of MELAS and MERRF. Amino Acids 2014, 46, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Aldini, G.; Carini, M.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med. 2006, 10, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Csordas, G.; Jowdy, C.; Schneider, T.; Csordas, N.; Wang, W.; Liu, Y.; Kohlhaas, M.; Meiser, M.; Bergem, S.; et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ. Res. 2012, 111, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. ER stress and its functional link to mitochondria: Role in cell survival and death. Cold Spring Harb. Perspect. Biol. 2011, 3, 1004424. [Google Scholar] [CrossRef] [PubMed]
- Bhandary, B.; Marahatta, A.; Kim, H.R.; Chae, H.J. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int. J. Mol. Sci. 2013, 14, 434–456. [Google Scholar] [CrossRef] [PubMed]
- Younce, C.W.; Burmeister, M.A.; Ayala, J.E. Exendin-4 attenuates high glucose-induced cardiomyocyte apoptosis via inhibition of endoplasmic reticulum stress and activation of SERCA2a. Am. J. Physiol. 2013, 304, C508–C518. [Google Scholar] [CrossRef] [PubMed]
- Groenendyk, J.; Sreenivasaiah, P.K.; Kim, D.H.; Agellon, L.B.; Michalak, M. Biology of endoplasmic reticulum stress in the heart. Circ. Res. 2010, 107, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Liu, X.R.; Cao, L.; Li, T.; Chen, L.L.; Yu, Y.Y.; Huang, W.J.; Liu, L.; Tan, X.Q. Propofol attenuates H2O2 induced oxidative stress and apoptosis via the mitochondria- and ER-medicated pathways in neonatal rat cardiomyocytes. Apoptosis 2017, 22, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Hulmi, J.J.; Hentila, J.; DeRuisseau, K.C.; Oliveira, B.M.; Papaioannou, K.G.; Autio, R.; Kujala, U.M.; Ritvos, O.; Kainulainen, H.; Korkmaz, A.; et al. Effects of muscular dystrophy, exercise and blocking activin receptor IIB ligands on the unfolded response and oxidative stress. Free Radic. Biol. Med. 2016, 99, 308–322. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.A.; Zhang, Z.; Svetlov, S.I.; Hayes, R.L.; Wang, K.K.; Larner, S.F. Calpain and caspase processing of caspase-12 contribute to the ER stress-induced cell death pathway in differentiated PC12 cells. Apoptosis 2010, 15, 1480–1493. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Yoshikawa, N.; Inui, T.; Miyazaki, N.; Schaffer, S.W.; Azuma, J. Tissue depletion of taurine accelerates skeletal muscle senescence and leads to early death in mice. PLoS ONE 2014, 9, e107409. [Google Scholar] [CrossRef] [PubMed]
- Jong, C.J.; Azuma, J.; Schaffer, S. Mechanism underlying the antioxidant activity of taurine: Prevention of mitochondrial oxidant production. Amino Acids 2012, 42, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Jong, C.J.; Ito, T.; Mozaffari, M.; Azuma, J.; Schaffer, S. Effect of beta-alanine treatment on mitochondrial tuarine level and 5-taurinomethyluridine content. J. Biomed. Sci. 2010, 17, S25. [Google Scholar] [CrossRef] [PubMed]
- Vartak, R.; Deng, J.; Fang, H.; Bai, Y. Redefining the roles of mitochondrial DNA-encoded subunits in respiratory complex I assembly. Biochim. Biophys. Acta 2015, 1852, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, S.W.; Shimada-Takaura, K.; Jong, C.J.; Ito, T.; Takahashi, K. Impaired energy metabolism of the taurine-deficient heart. Amino Acids 2016, 48, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Scheubel, R.J.; Tostlebe, M.; Simm, A.; Rohrbach, S.; Prondzinsky, R.; Gellerich, F.N.; Silber, R.E.; Holtz, J. Dysfunction of mitochondrial respiratory chain complex I in human failing myocardium is not due to disturbed mitochondrial gene expression. J. Am. Coll. Cardiol. 2002, 40, 2174–2181. [Google Scholar] [CrossRef]
- Sanders, L.H.; Greenamyre, J.T. Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radic. Biol. Med. 2013, 62, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bleier, L.; Wittig, I.; Heide, H.; Steger, M.; Brandt, U.; Drose, S. Generator-specific targets of mitochondrial reactive oxygen species. Free Radic. Biol. Med. 2015, 78, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puiq, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, T.; Newmeyer, D.D. Bcl-2 family proteins and the role of mitochondria in apoptosis. Curr. Opin. Cell Biol. 2003, 15, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Wu, D.; Chen, W.; Yan, Z.; Shi, Y. Proteolytic processing of the caspase-9 zymogen is required for apoptosome-mediated activation of caspase-9. J. Biol. Chem. 2013, 288, 15142–15147. [Google Scholar] [CrossRef] [PubMed]
- Montessuit, S.; Somasekharan, S.P.; Terrones, O.; Lucken-Ardjomande, S.; Herzig, S.; Schwarzenbacher, R.; Manstein, D.J.; Bossy-Wetzel, E.; Basanez, G.; Meda, P.; et al. Membrane remodeling induced by dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 2010, 142, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Kashnareva, Y.; Andreyev, A.Y.; Kuwana, T.; Newmeyer, D.D. Bax activation initiates the assembly of a multimeric catalyst that facilitates Bax pore formation in mitochondrial outer membranes. PLoS Biol. 2012, 10, e1001394. [Google Scholar] [CrossRef] [PubMed]
- Jong, C.J.; Ito, T.; Schaffer, S.W. The ubiquitin-proteasome system and autophagy are defective in the taurine-deficient heart. Amino Acids 2015, 47, 2609–2622. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Prentice, H.; Price, A.L.; Wu, J.Y. Beneficial effect of taurine on hypoxia- and glutamate-induced endoplasmic reticulum stress pathways in primary neuronal culture. Amino Acids 2012, 43, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Sinha, K.; Banerjee, S.; Sil, P.C. Taurine protects cisplatin-induced cardiotoxicity by modulating inflammatory and endoplasmic reticulum stress responses. Biofactors 2016, 42, 647–664. [Google Scholar] [CrossRef] [PubMed]
- Prentice, H.; Modi, J.P.; Wu, J.-Y. Mechanisms of neuronal protection against excitotoxicity, endoplasmic reticulum stress, and mitochondrial dysfunction in stroke and neurodegenerative diseases. Oxid. Med. Cell. Longev. 2015, 2015, 964518. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Chen, Q.; Hoppel, C.L. Mitochondrial metabolism in aging heart. Circ. Res. 2016, 118, 1593–1611. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Martinon, F.; Rodriguez, D.; Glimcher, L.H. The unfolded protein response: Integrating stress signals through the stress sensor IRE1α. Physiol. Rev. 2011, 91, 1219–1243. [Google Scholar] [CrossRef] [PubMed]









© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jong, C.J.; Ito, T.; Prentice, H.; Wu, J.-Y.; Schaffer, S.W. Role of Mitochondria and Endoplasmic Reticulum in Taurine-Deficiency-Mediated Apoptosis. Nutrients 2017, 9, 795. https://doi.org/10.3390/nu9080795
Jong CJ, Ito T, Prentice H, Wu J-Y, Schaffer SW. Role of Mitochondria and Endoplasmic Reticulum in Taurine-Deficiency-Mediated Apoptosis. Nutrients. 2017; 9(8):795. https://doi.org/10.3390/nu9080795
Chicago/Turabian StyleJong, Chian Ju, Takashi Ito, Howard Prentice, Jang-Yen Wu, and Stephen W. Schaffer. 2017. "Role of Mitochondria and Endoplasmic Reticulum in Taurine-Deficiency-Mediated Apoptosis" Nutrients 9, no. 8: 795. https://doi.org/10.3390/nu9080795
APA StyleJong, C. J., Ito, T., Prentice, H., Wu, J. -Y., & Schaffer, S. W. (2017). Role of Mitochondria and Endoplasmic Reticulum in Taurine-Deficiency-Mediated Apoptosis. Nutrients, 9(8), 795. https://doi.org/10.3390/nu9080795