Distal Colon Motor Dysfunction in Mice with Chronic Kidney Disease: Putative Role of Uremic Toxins


Abstract
:1. Introduction
2. Results
2.1. Mice Fed an Adenin Diet Developed Renal Failure
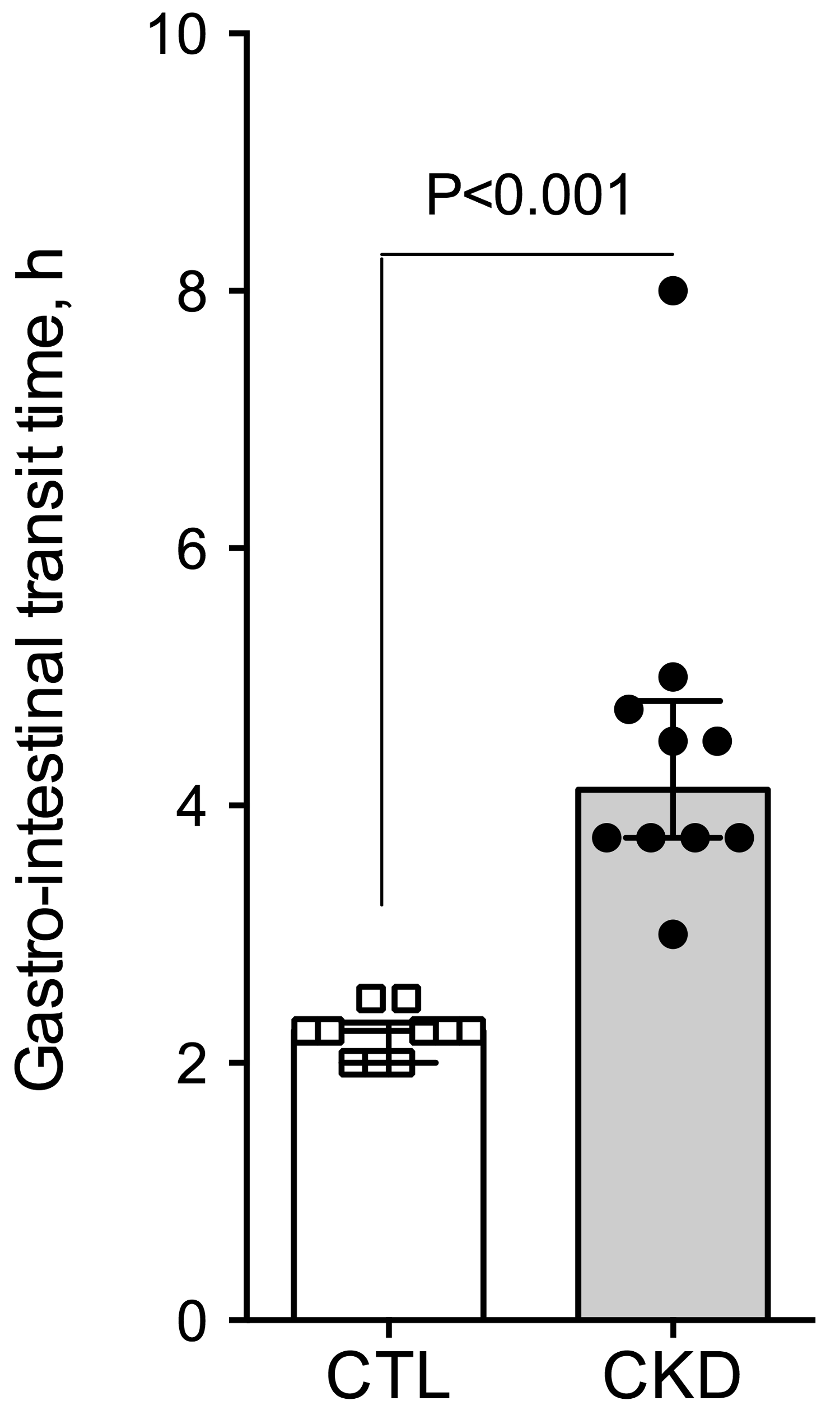
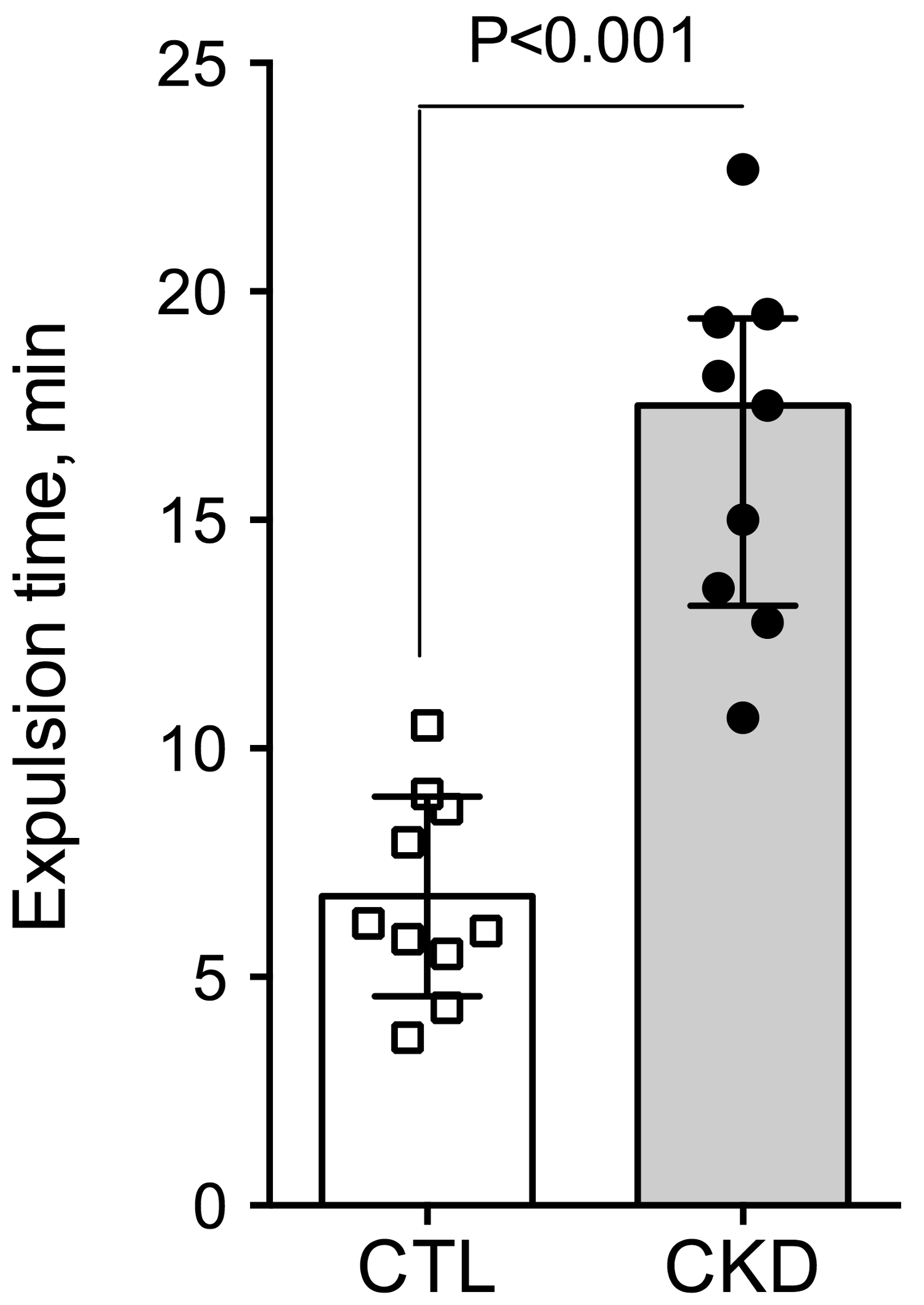
2.2. Effect of Uremia on Gastrointestinal Transit Time
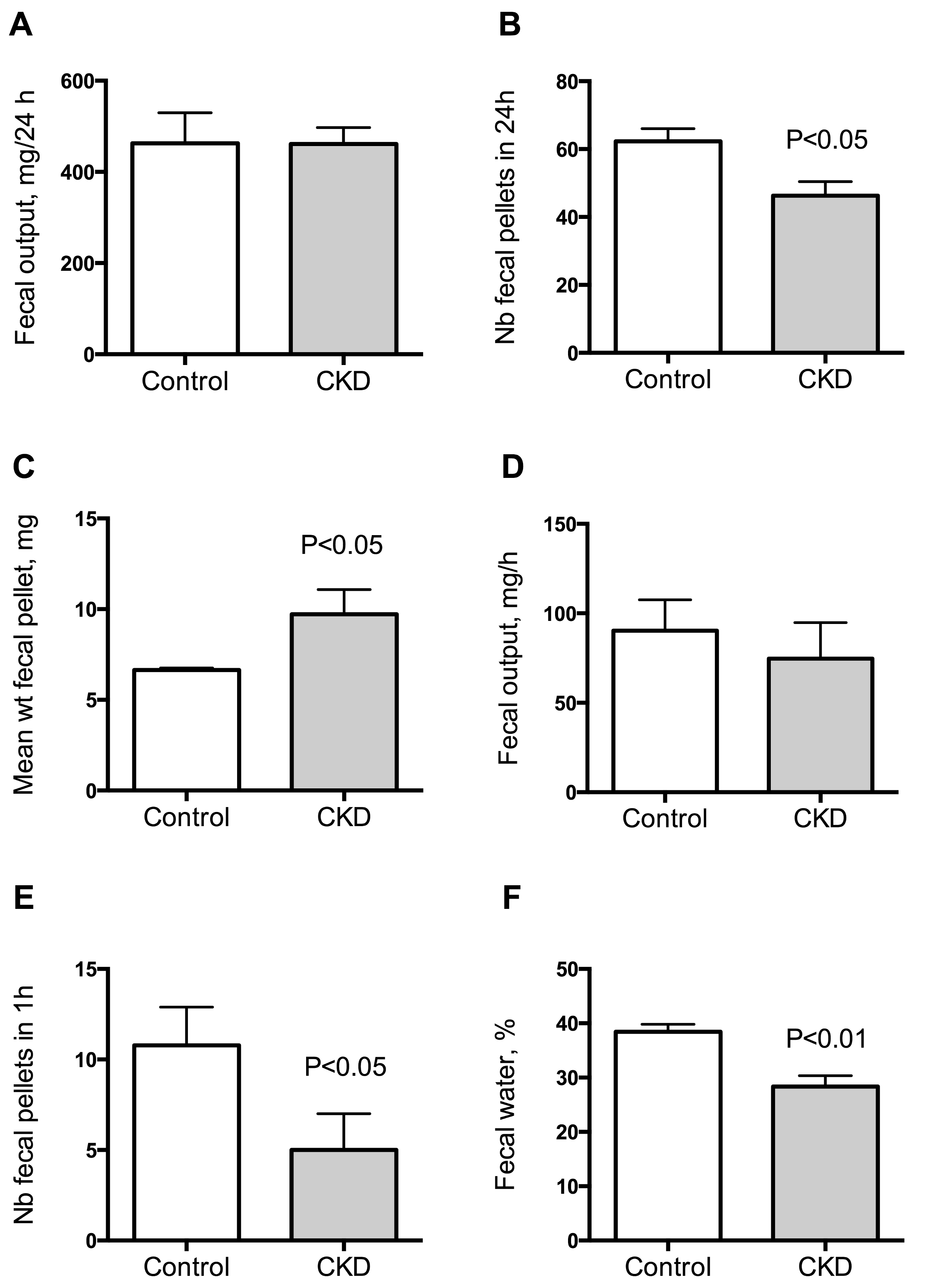
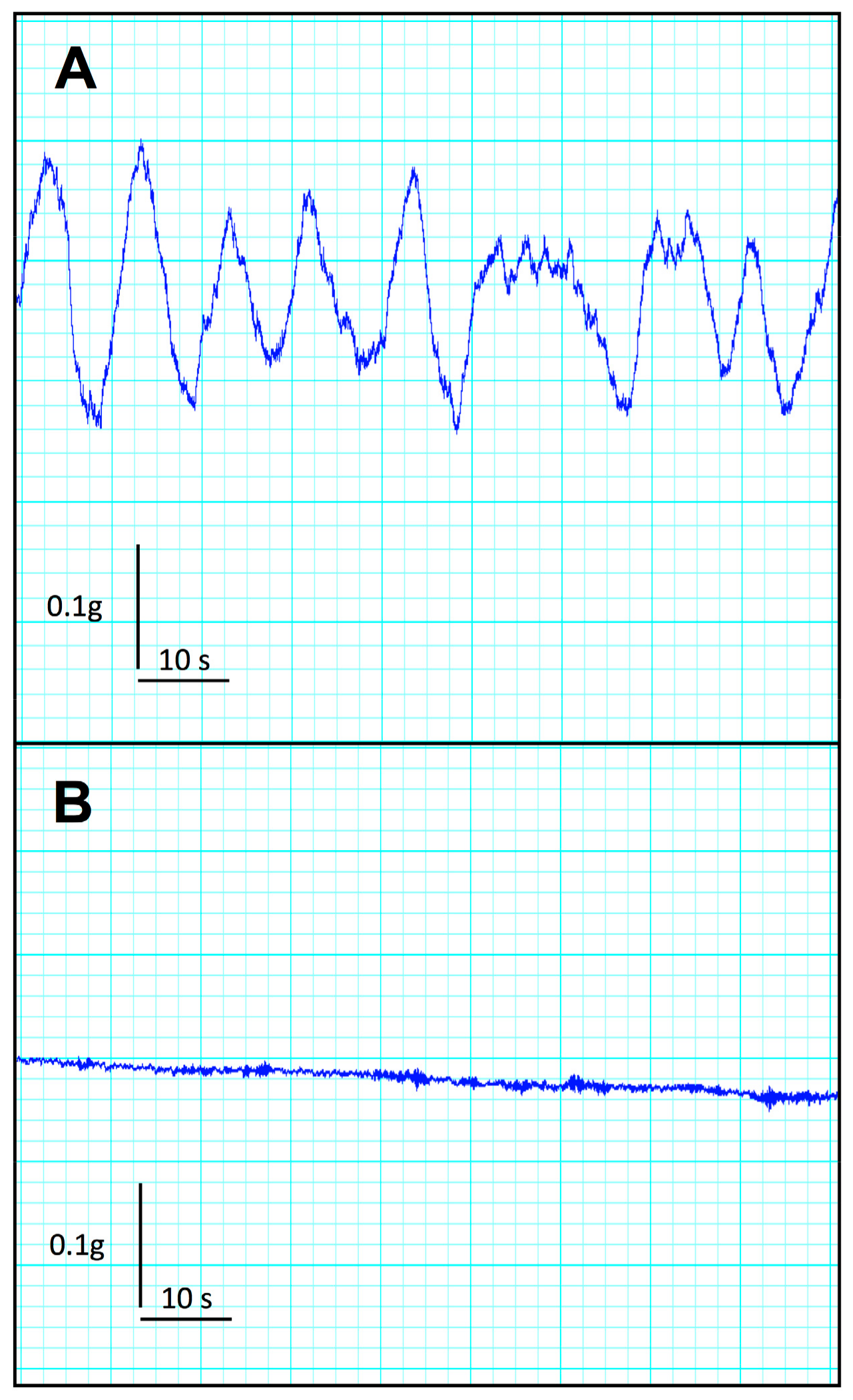
2.3. Effect of Uremia on Duodenum and Colon Motility
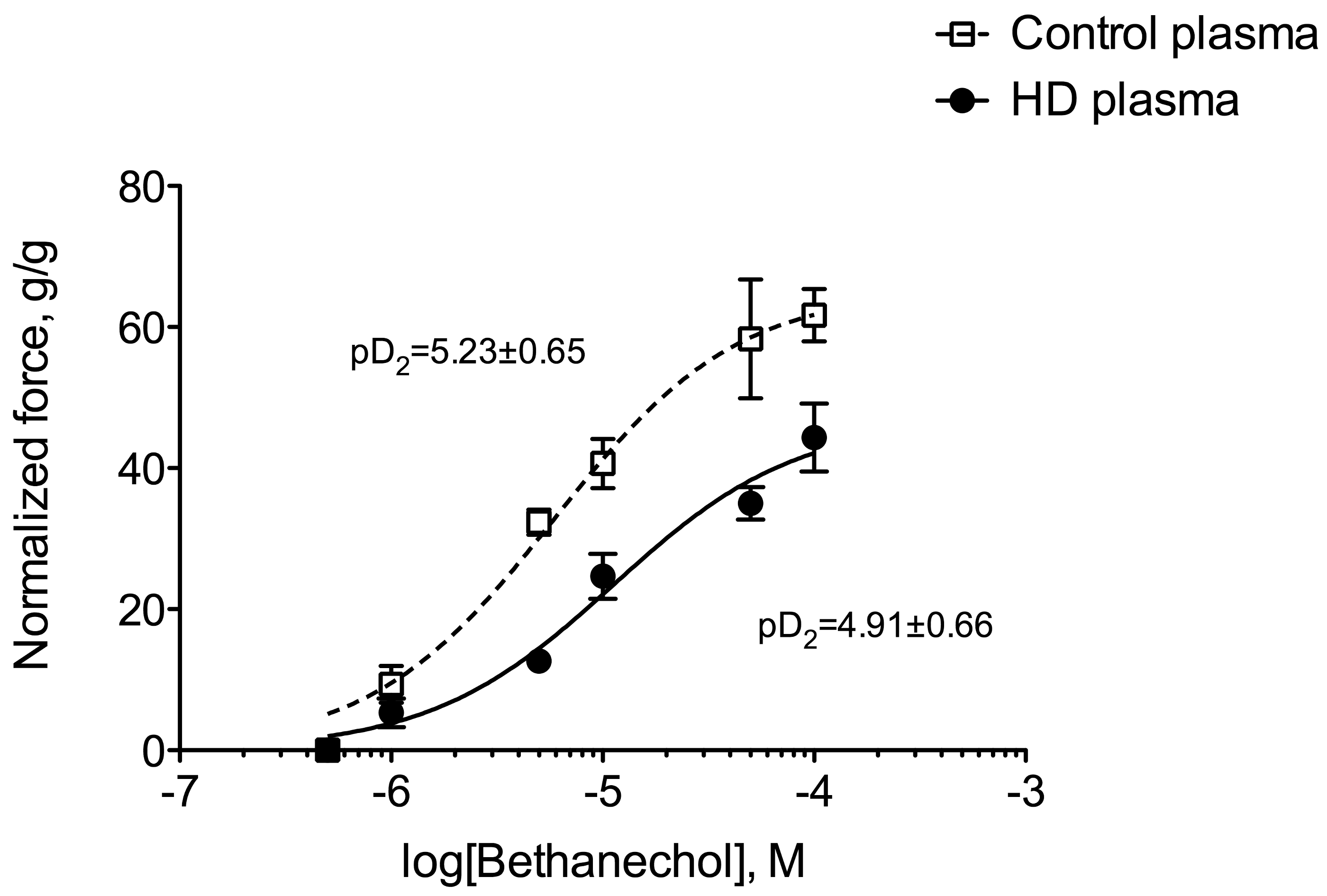
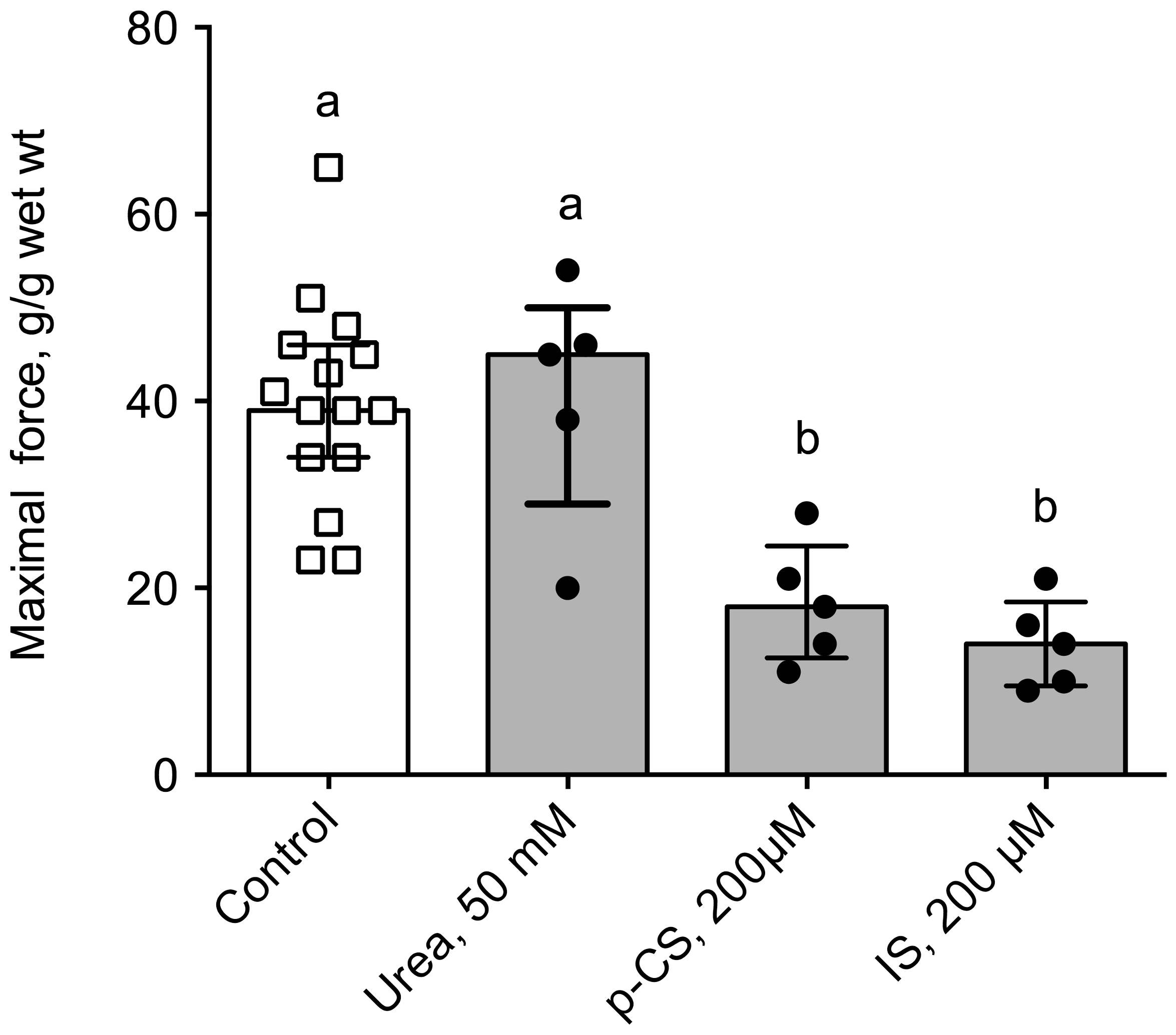
2.4. Uremic Plasma and Uremic Toxins Induce Gut Dysmotility
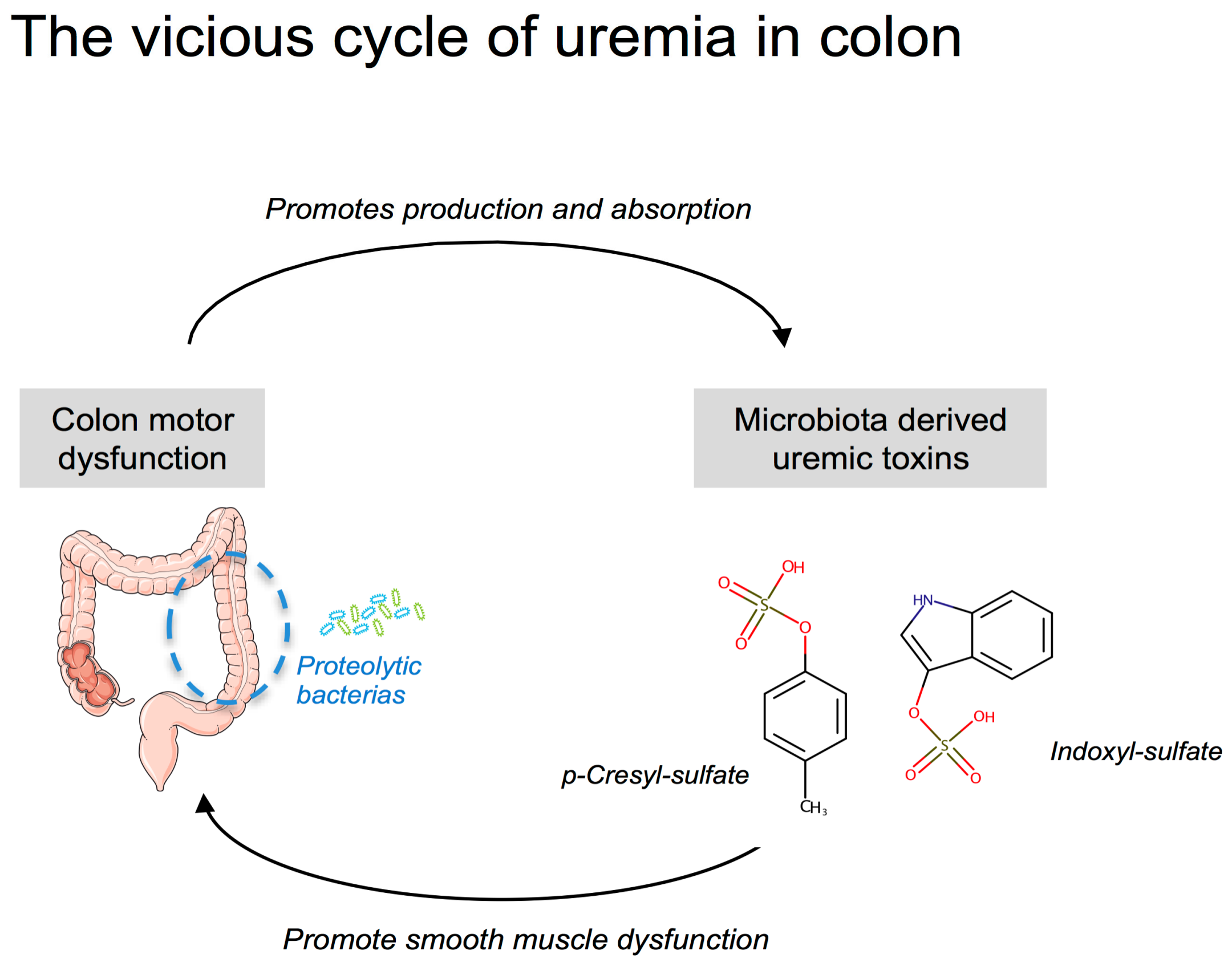
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Chemicals and Reagents
5.2. Patients
5.3. Animals
5.4. Measurement of Total Gastrointestinal Transit Time
5.5. Urine Collection and Biochemical Measurements
5.6. Fecal Water Content
5.7. Measurement of Colonic Motility In Vivo
5.8. Sacrifice and Tissue Dissection
5.9. Isolated Duodenum and Colon
5.10. Statistics
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Cano, A.E.; Neil, A.K.; Kang, J.Y.; Barnabas, A.; Eastwood, J.B.; Nelson, S.R.; Hartley, I.; Maxwell, D. Gastrointestinal symptoms in patients with end-stage renal disease undergoing treatment by hemodialysis or peritoneal dialysis. Am. J. Gastroenterol. 2007, 102, 1990–1997. [Google Scholar] [CrossRef] [PubMed]
- Doğan, I.; Unal, S.; Sindel, S.; Tunçer, C.; Arinsoy, T.; Bali, M.; Kandilci, U.; Hasanoğlu, E. Esophageal motor dysfunction in chronic renal failure. Nephron. 1996, 72, 346–347. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Mellow, M.H.; Southmayd, L.; Pan, J.; Chen, J.D. Impaired gastric myoelectrical activity in patients with chronic renal failure. Dig. Dis. Sci. 1997, 42, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Hirako, M.; Kamiya, T.; Misu, N.; Kobayashi, Y.; Adachi, H.; Shikano, M.; Matsuhisa, E.; Kimura, G. Impaired gastric motility and its relationship to gastrointestinal symptoms in patients with chronic renal failure. J. Gastroenterol. 2005, 40, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Furgała, A.; Błaut-Kadzielska, U.; Stojakowska, M.; Dobrek, Ł.; Mazur, M.; Machowska, A.; Thor, P.J. Gastric dysfunction in dialysed patients with chronic renal failure. Folia Med. Cracov. 2012, 52, 39–55. [Google Scholar] [PubMed]
- Musola, R.; Franzin, G.; Mora, R.; Manfrini, C. Prevalence of gastroduodenal lesions in uremic patients undergoing dialysis and after renal transplantation. Gastrointest. Endosc. 1984, 30, 343–346. [Google Scholar] [CrossRef]
- Strid, H.; Simrén, M.; Stotzer, P.-O.; Ringström, G.; Abrahamsson, H.; Björnsson, E.S. Patients with chronic renal failure have abnormal small intestinal motility and a high prevalence of small intestinal bacterial overgrowth. Digestion 2003, 67, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.J.; Chang, C.S.; Cheng, C.H.; Chen, C.H.; Lee, W.C.; Hsu, Y.H.; Shu, K.H.; Tang, M.J. Colonic transit time in long-term dialysis patients. Am. J. Kidney Dis. 2004, 44, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.Y. The gastrointestinal tract in uremia. Dig. Dis. Sci. 1993, 38, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, A.M. Gastrointestinal function in chronic renal failure. Pediatr. Nephrol. 1995, 9, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Nagib, E.M.; El-Sayed, M.H.; Ahmed, M.A.; Youssef, M.H. Intestinal motility in acute uremia and effects of erythropoietin. Saudi Med. J. 2012, 33, 500–507. [Google Scholar] [PubMed]
- Lefebvre, H.P.; Ferré, J.P.; Watson, A.D.; Brown, C.A.; Serthelon, J.P.; Laroute, V.; Concordet, D.; Toutain, P.L. Small bowel motility and colonic transit are altered in dogs with moderate renal failure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R230–R238. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.G.; Wang, Y.; Yuan, H.Z.; Zhou, J.P.; Wang, L.; Liu, X.D.; Ma, F.; Zhang, J. Effects of chronic renal failure on gastrointestinal motility: A study on the changes of gastric emptying, small intestinal transit, interdigestive myoelectric complex, and fecal water content. Renal Fail. 2011, 33, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Da Graça, J.R.V.; Parente, C.C.; Fiúza, R.F.; da Silva, P.A.F.; Mota, B.T.; Salles, L.D.; de Souza Silva, C.M.; da Silva, M.T.B.; de Oliveira, R.B.; Dos Santos, A.A. Subtotal nephrectomy inhibits the gastric emptying of liquid in awake rats. Physiol. Rep. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Hegbrant, J.; Thysell, H.; Ekman, R. Plasma levels of gastrointestinal regulatory peptides in patients receiving maintenance hemodialysis. Scand. J. Gastroenterol. 1991, 26, 599–604. [Google Scholar] [CrossRef] [PubMed]
- McLeod, R.S.; Track, N.S.; Reynolds, L.E. Elevated plasma motilin concentrations in chronic renal disease. Can. Med. Assoc. J. 1979, 121, 268–273. [Google Scholar] [PubMed]
- Piga, M.; Altieri, P.; Floris, A.; Barraca, A.; Tagleri, G.; Madeddu, G.; Bolasco, F.; Gervasi, F.; Piras, M.R.; Serra, A.R. Vasoactive intestinal polypeptide (VIP) plasma levels in chronic renal failure. J. Nucl. Med. Allied Sci. 1984, 28, 77–80. [Google Scholar] [PubMed]
- Shima, K.; Shin, S.; Tanaka, A.; Hashimura, E.; Nishino, T.; Imagawa, K.; Kumahara, Y.; Yanaihara, N. Heterogeneity of plasma motilin in patients with chronic renal failure. Horm. Metab. Res. 1980, 12, 328–331. [Google Scholar] [CrossRef] [PubMed]
- Sirinek, K.R.; O’Dorisio, T.M.; Gaskill, H.V.; Levine, B.A. Chronic renal failure: Effect of hemodialysis on gastrointestinal hormones. Am. J. Surg. 1984, 148, 732–735. [Google Scholar] [CrossRef]
- Chou, Y.H.; Tsai, T.J. Autonomic dysfunction in chronic kidney disease: An old problem in a new era. J. Formos. Med. Assoc. 2016, 115, 687–688. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. European Uremic Toxin Work Group. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.; Glorieux, G.; Thornalley, P.; Schepers, E.; Meert, N.; Jankowski, J.; Jankowski, V.; Argiles, A.; Anderstam, B.; Brunet, P.; et al. Review on uraemic toxins III: Recommendations for handling uraemic retention solutes in vitro--towards a standardized approach for research on uraemia. Nephrol. Dial. Transplant. 2007, 22, 3381–3390. [Google Scholar] [CrossRef] [PubMed]
- Yokozawa, T.; Zheng, P.D.; Oura, H.; Koizumi, F. Animal model of adenine-induced chronic renal failure in rats. Nephron. 1986, 44, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Yokozawa, T.; Oura, H.; Okada, T. Metabolic effects of dietary purine in rats. J. Nutr. Sci. Vitaminol. 1982, 28, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Kaneko, Y.; Yawata, T.; Uyama, H.; Ozono, S.; Motomiya, Y.; Hirao, Y. Reversibility of adenine-induced renal failure in rats. Clin. Exp. Nephrol. 1999, 3, 82–88. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Yuan, J.; Rahimi, A.; Ni, Z.; Said, H.; Subramanian, V.S. Disintegration of colonic epithelial tight junction in uremia: A likely cause of CKD-associated inflammation. Nephrol. Dial. Transplant. 2012, 27, 2686–2693. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Fukuda, S.; Shima, H.; Hirayama, A.; Akiyama, Y.; Takeuchi, Y.; Fukuda, N.N.; Suzuki, T.; Suzuki, C.; Yuri, A.; et al. Alteration of the Intestinal Environment by Lubiprostone Is Associated with Amelioration of Adenine-Induced CKD. J. Am. Soc. Nephrol. 2015, 26, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.J.; Cai, X.D.; Xing, J.; Zhong, G.H.; Chen, J.D.Z. Circulating motilin, ghrelin, and GLP-1 and their correlations with gastric slow waves in patients with chronic kidney disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 313, R149–R157. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Luo, S.; Liu, Y. Effects of changes of plasma motilin level on the motility of gallbladder in patients with chronic renal failure. Zhonghua Nei Ke Za Zhi 1996, 35, 86–88. [Google Scholar] [PubMed]
- Taylor, I.L.; Sells, R.A.; McConnell, R.B.; Dockray, G.J. Serum gastrin in patients with chronic renal failure. Gut. 1980, 21, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Hausberg, M.; Kosch, M.; Harmelink, P.; Barenbrock, M.; Hohage, H.; Kisters, K.; Dietl, K.H.; Rahn, K.H. Sympathetic nerve activity in end-stage renal disease. Circulation. 2002, 106, 1974–1979. [Google Scholar] [CrossRef] [PubMed]
- Adachi, H.; Kamiya, T.; Hirako, M.; Misu, N.; Kobayashi, Y.; Shikano, M.; Matsuhisa, E.; Kataoka, H.; Sasaki, M.; Ohara, H.; et al. Improvement of gastric motility by hemodialysis in patients with chronic renal failure. J. Smooth Muscle Res. 2007, 43, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Baurmeister, U.; Brunet, P.; Cohen, G.; Glorieux, G.; Jankowski, J. European Uremic Toxin Work Group. A bench to bedside view of uremic toxins. J. Am. Soc. Nephrol. 2008, 19, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Meert, N.; Schepers, E.; Glorieux, G.; Argiles, A.; Brunet, P.; Cohen, G.; Drüeke, T.; Mischak, H.; Spasovski, G.; et al. Review on uraemic solutes II—variability in reported concentrations: Causes and consequences. Nephrol. Dial. Transplant. 2007, 22, 3115–3121. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.L.; Vaziri, N.D. Urea, a true uremic toxin: The empire strikes back. Clin. Sci. 2017, 131, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Yuan, J.; Norris, K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am. J. Nephrol. 2013, 37, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Miyamoto, Y.; Enoki, Y.; Ishima, Y.; Kadowaki, D.; Kotani, S.; Nakajima, M.; Tanaka, M.; Matsushita, K.; Mori, Y.; et al. p-Cresyl sulfate, a uremic toxin, causes vascular endothelial and smooth muscle cell damages by inducing oxidative stress. Pharmacol. Res. Perspect. 2015, 3, e00092. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Tsuruoka, S.; Ioka, T.; Ando, H.; Ito, C.; Akimoto, T.; Fujimura, A.; Asano, Y.; Kusano, E. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int. 2006, 69, 1780–1785. [Google Scholar] [CrossRef] [PubMed]
- Muteliefu, G.; Enomoto, A.; Niwa, T. Indoxyl sulfate promotes proliferation of human aortic smooth muscle cells by inducing oxidative stress. J. Ren. Nutr. 2009, 19, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Muteliefu, G.; Enomoto, A.; Jiang, P.; Takahashi, M.; Niwa, T. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol. Dial. Transplant. 2009, 24, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, J.; Shen, Z.; Gu, Y.; Xu, L.; Hu, J.; Zhang, X.; Ding, X. Indoxyl sulfate accelerates vascular smooth muscle cell calcification via microRNA-29b dependent regulation of Wnt/β-catenin signaling. Toxicol. Lett. 2018, 284, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.C.; Dinatale, B.C.; Murray, I.A.; Flaveny, C.A.; Liu, Q.; Laurenzana, E.M.; Lin, J.M.; Strom, S.C.; Omiecinski, C.J.; Amin, S.; et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry. 2010, 49, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Koppe, L.; Pillon, N.J.; Vella, R.E.; Croze, M.L.; Pelletier, C.C.; Chambert, S.; Massy, Z.; Glorieux, G.; Vanholder, R.; Dugenet, Y.; et al. p-Cresyl sulfate promotes insulin resistance associated with CKD. J. Am. Soc. Nephrol. 2013, 24, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Mathialahan, T.; Sandle, G.I. Dietary potassium and laxatives as regulators of colonic potassium secretion in end-stage renal disease. Nephrol. Dial. Transplant. 2003, 18, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Aronov, P.A.; Luo, F.J.-G.; Plummer, N.S.; Quan, Z.; Holmes, S.; Hostetter, T.H.; Meyer, T.W. Colonic contribution to uremic solutes. J. Am. Soc. Nephrol. 2011, 22, 1769–1776. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Fukuda, S.; Mukawa, C.; Yuri, A.; Kanemitsu, Y.; Matsumoto, Y.; Akiyama, Y.; Fukuda, N.N.; Tsukamoto, H.; Asaji, K.; et al. Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS-based metabolomics approach. Kidney Int. 2017, 92, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Einheber, A.; Carter, D. The role of the microbial flora in uremia. I. Survival times of germfree, limited-flora, and conventionalized rats after bilateral nephrectomy and fasting. J. Exp. Med. 1966, 123, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Evenepoel, P.; Meijers, B.K.I.; Bammens, B.R.M.; Verbeke, K. Uremic toxins originating from colonic microbial metabolism. Kidney Int. Suppl. 2009, 76, S12–S19. [Google Scholar] [CrossRef] [PubMed]
- Poesen, R.; Meijers, B.; Evenepoel, P. The colon: An overlooked site for therapeutics in dialysis patients. Semin. Dial. 2013, 26, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Glorieux, G. The intestine and the kidneys: A bad marriage can be hazardous. Clin. Kidney J. 2015, 8, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H.; Hill, M.J.; Bone, E.S.; Branch, W.J.; Jenkins, D.J. The effect of meat protein and dietary fiber on colonic function and metabolism. II. Bacterial metabolites in feces and urine. Am. J. Clin. Nutr. 1979, 32, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Feigenbaum, J.; Neuberg, C.A. Simplified Method for the Preparation of Aromatic Sulfuric Acid Esters. J. Am. Chem. Soc. 1941, 63, 3529–3530. [Google Scholar] [CrossRef]
- Dey, N.; Wagner, V.E.; Blanton, L.V.; Cheng, J.; Fontana, L.; Haque, R.; Ahmed, T.; Gordon, J.I. Regulators of gut motility revealed by a gnotobiotic model of diet-microbiome interactions related to travel. Cell. 2015, 163, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M.; Linden, D.R. Measurement of Gastrointestinal and Colonic Motor Functions in Humans and Animals. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 412–428. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Luque-de Leon, E.; Kost, L.J.; Sarr, M.G.; Phillips, S.F. Duodenal motility in fasting dogs: Humoral and neural pathways mediating the colonic brake. Am. J. Physiol. 1998, 274, G192–G195. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control (n = 16) | CKD (n = 15) | p-Value | |||||
|---|---|---|---|---|---|---|---|
| Biometry | |||||||
| Body weight, g | 20.6 | ± | 0.2 | 15.9 | ± | 0.8 | <0.001 |
| Kidney, mg/10 g bw | 126 | ± | 9 | 77 | ± | 12 | <0.001 |
| Heart, mg/10 g bw | 53 | ± | 7 | 63 | ± | 11 | 0.023 |
| Liver, mg/10 g bw | 408 | ± | 13 | 415 | ± | 50 | 0.529 |
| Intestine, mg/10 g bw | 335 | ± | 20 | 385 | ± | 35 | <0.001 |
| Colon, 10 mg bw | 53 | ± | 9 | 65 | ± | 14 | 0.065 |
| Biochemistry | |||||||
| Plasma urea, mmol/L | 8.6 | ± | 0.8 | 27.9 | ± | 4.0 | <0.001 |
| Proteinuria, mg/24 h | 7.9 | ± | 6.9 | 36.0 | ± | 21.8 | 0.100 |
| Control (n = 6) | CKD (n = 5) | p-Value | |||||
|---|---|---|---|---|---|---|---|
| Duodenum | |||||||
| Frequency of contraction, min−1 | 36.3 | ± | 4.5 | 30.7 | ± | 4.7 | 0.583 |
| Average duration of contraction, s | 1.8 | ± | 0.3 | 2.3 | ± | 0.4 | 0.355 |
| Average force of contraction, g/g wet wt | 2.4 | ± | 0.6 | 1.8 | ± | 0.6 | 0.485 |
| Maximal force of contraction, g/g wet wt * | 6.6 | ± | 3.3 | 6.5 | ± | 1.4 | 0.920 |
| Motility index, AU | 8.8 | ± | 0.4 | 7.0 | ± | 1.5 | 0.288 |
| Descending colon | |||||||
| Frequency of contraction, min−1 | 6.8 | ± | 3.5 | <1 | - | ||
| Average duration of contraction, s | 14.1 | ± | 6.7 | nd | - | ||
| Average force of contraction, g/g wet wt | 3.7 | ± | 1.4 | 0.07 | ± | 0.03 | 0.016 |
| Maximal force of contraction, g/g wet wt * | 35.5 | ± | 11.2 | 0.9 | ± | 0.6 | <0.001 |
| Motility index, AU | 8.8 | ± | 1.1 | nd | - | ||
| Control (n = 11) | Hemodialysis (n = 19) | p-Value | |||||
|---|---|---|---|---|---|---|---|
| Gender, M/F | 6/5 | 13/6 | 0.696 | ||||
| Age, y | 49.0 | ± | 12.9 | 62.4 | ± | 14.6 | 0.017 |
| Dialysis vintage, y | n/a | 3.7 | ± | 2.6 | - | ||
| Creatinine, µM | 80 | ± | 32 | 818 | ± | 402 | <0.001 |
| eGFR, mL/min.1.73m2 | 94 | ± | 29 | <15 | - | ||
| Urea, mM | 6.6 | ± | 2.7 | 20.7 | ± | 8.0 | 0.007 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoibian, E.; Florens, N.; Koppe, L.; Vidal, H.; Soulage, C.O. Distal Colon Motor Dysfunction in Mice with Chronic Kidney Disease: Putative Role of Uremic Toxins. Toxins 2018, 10, 204. https://doi.org/10.3390/toxins10050204
Hoibian E, Florens N, Koppe L, Vidal H, Soulage CO. Distal Colon Motor Dysfunction in Mice with Chronic Kidney Disease: Putative Role of Uremic Toxins. Toxins. 2018; 10(5):204. https://doi.org/10.3390/toxins10050204
Chicago/Turabian StyleHoibian, Elsa, Nans Florens, Laetitia Koppe, Hubert Vidal, and Christophe O. Soulage. 2018. "Distal Colon Motor Dysfunction in Mice with Chronic Kidney Disease: Putative Role of Uremic Toxins" Toxins 10, no. 5: 204. https://doi.org/10.3390/toxins10050204
APA StyleHoibian, E., Florens, N., Koppe, L., Vidal, H., & Soulage, C. O. (2018). Distal Colon Motor Dysfunction in Mice with Chronic Kidney Disease: Putative Role of Uremic Toxins. Toxins, 10(5), 204. https://doi.org/10.3390/toxins10050204