Basics of Antibody Phage Display Technology

 ,
, 
Abstract
:1. Introduction
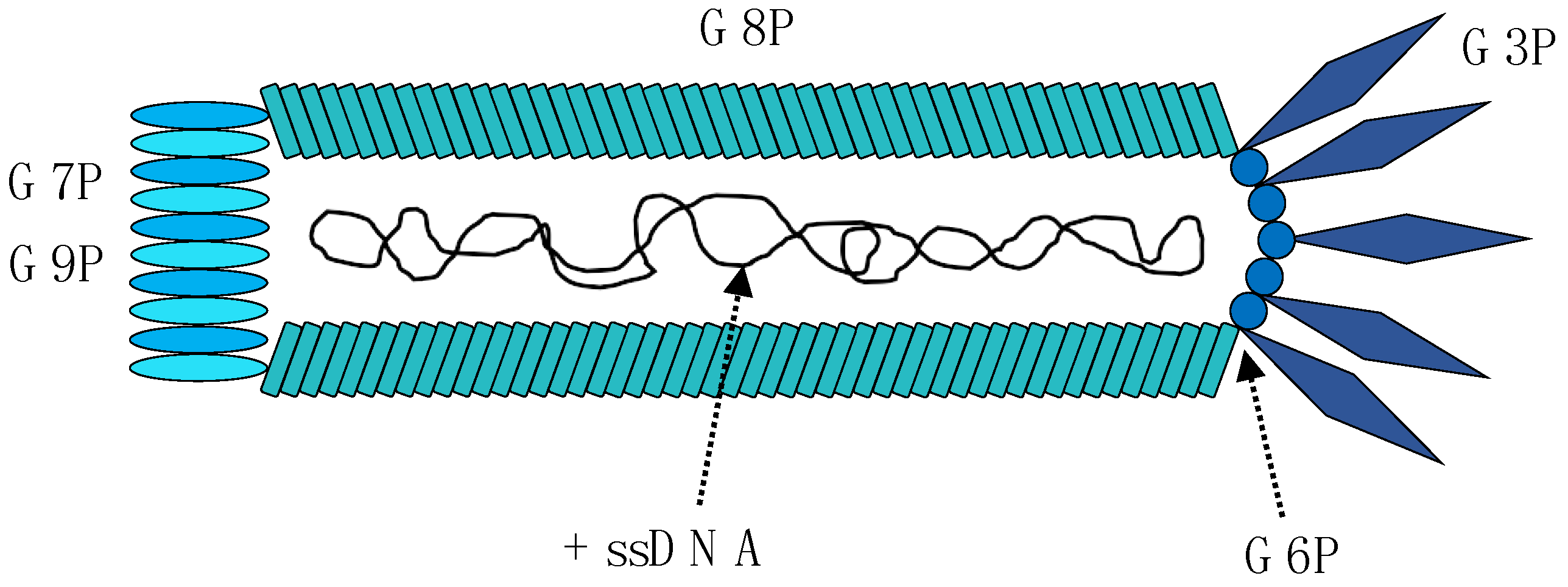
2. The M13 Bacteriophage
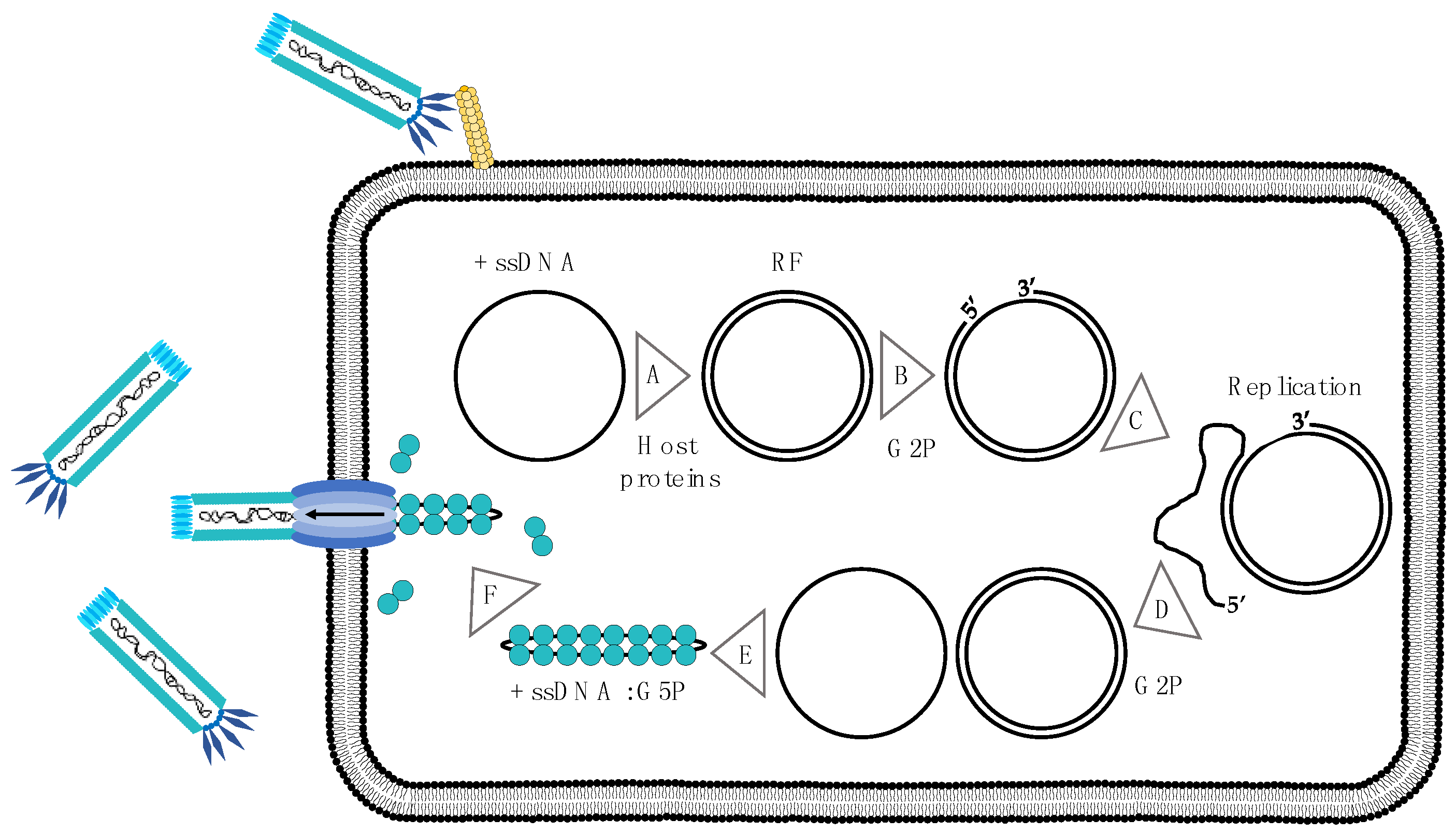
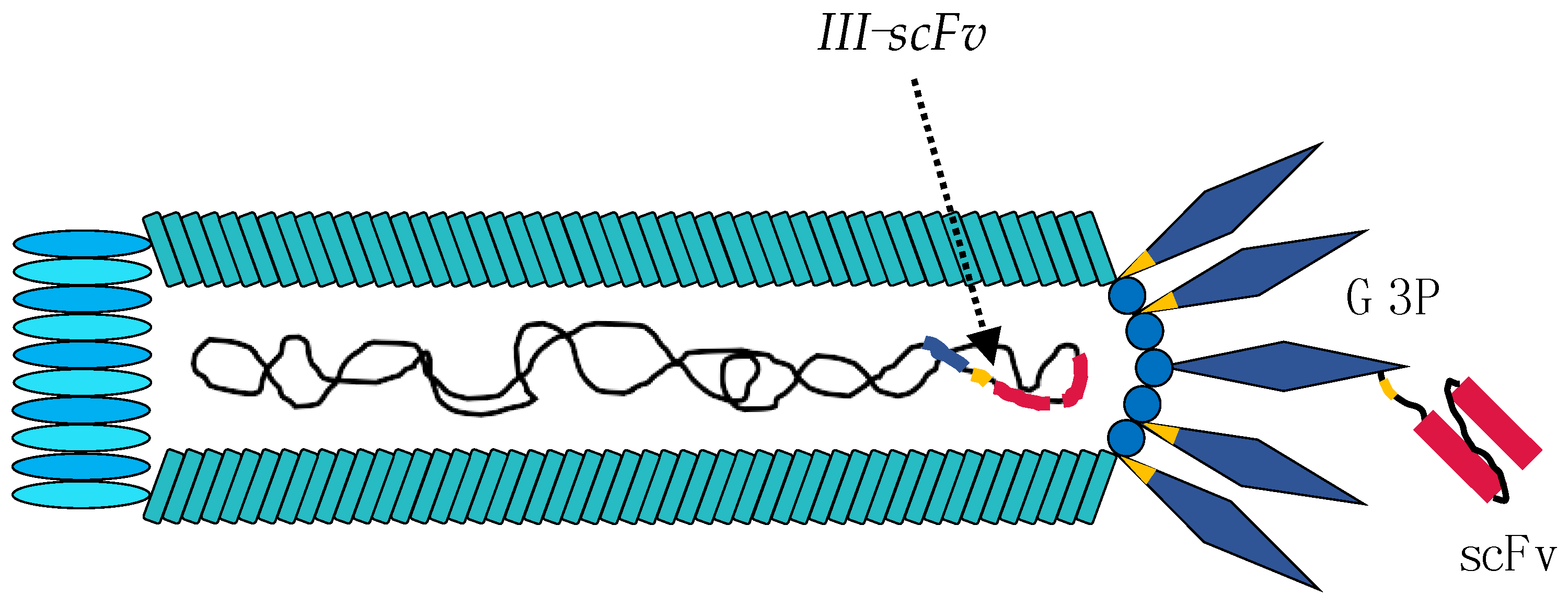
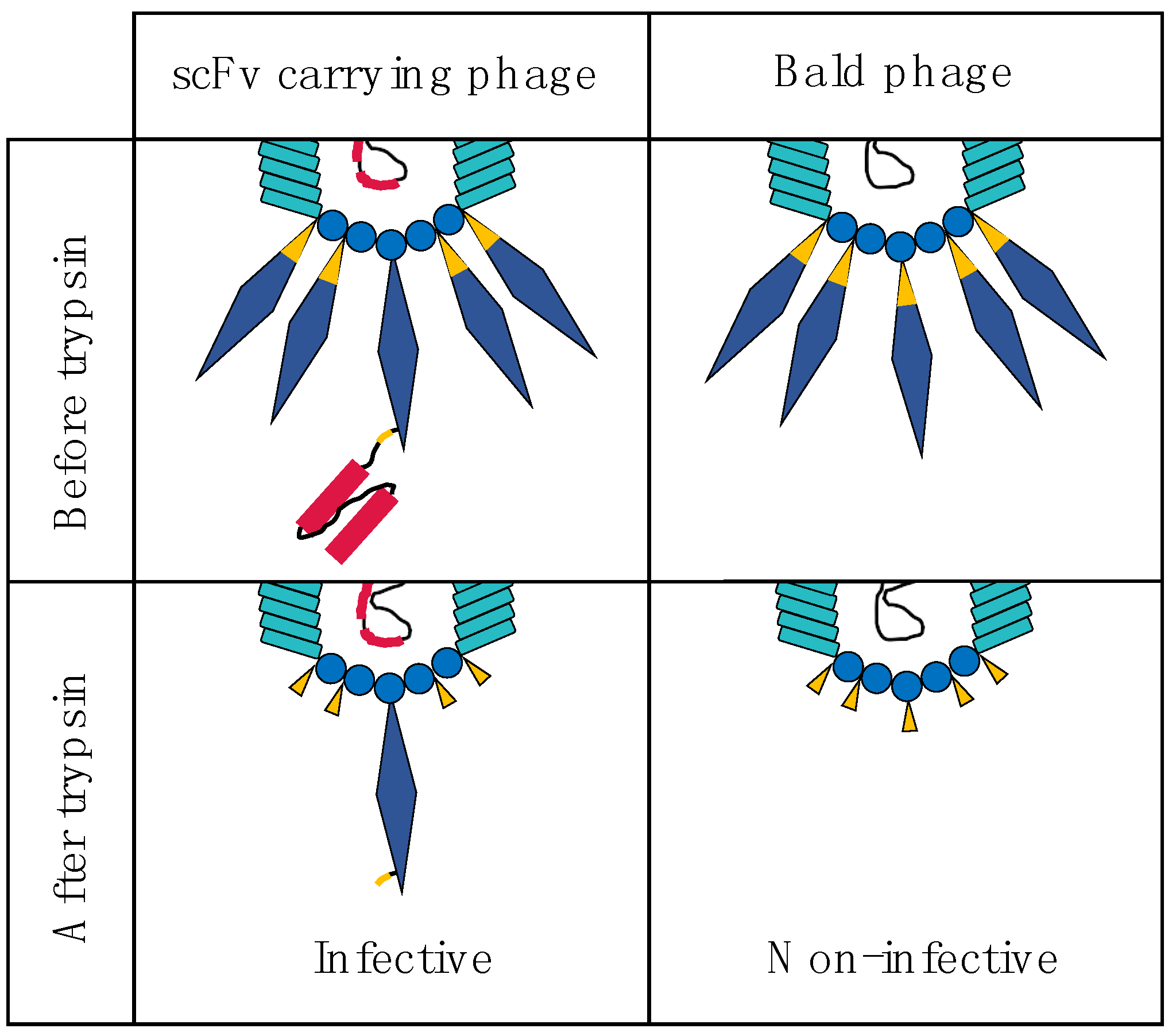
3. Using the M13 Phage as a Tool in Antibody Discovery
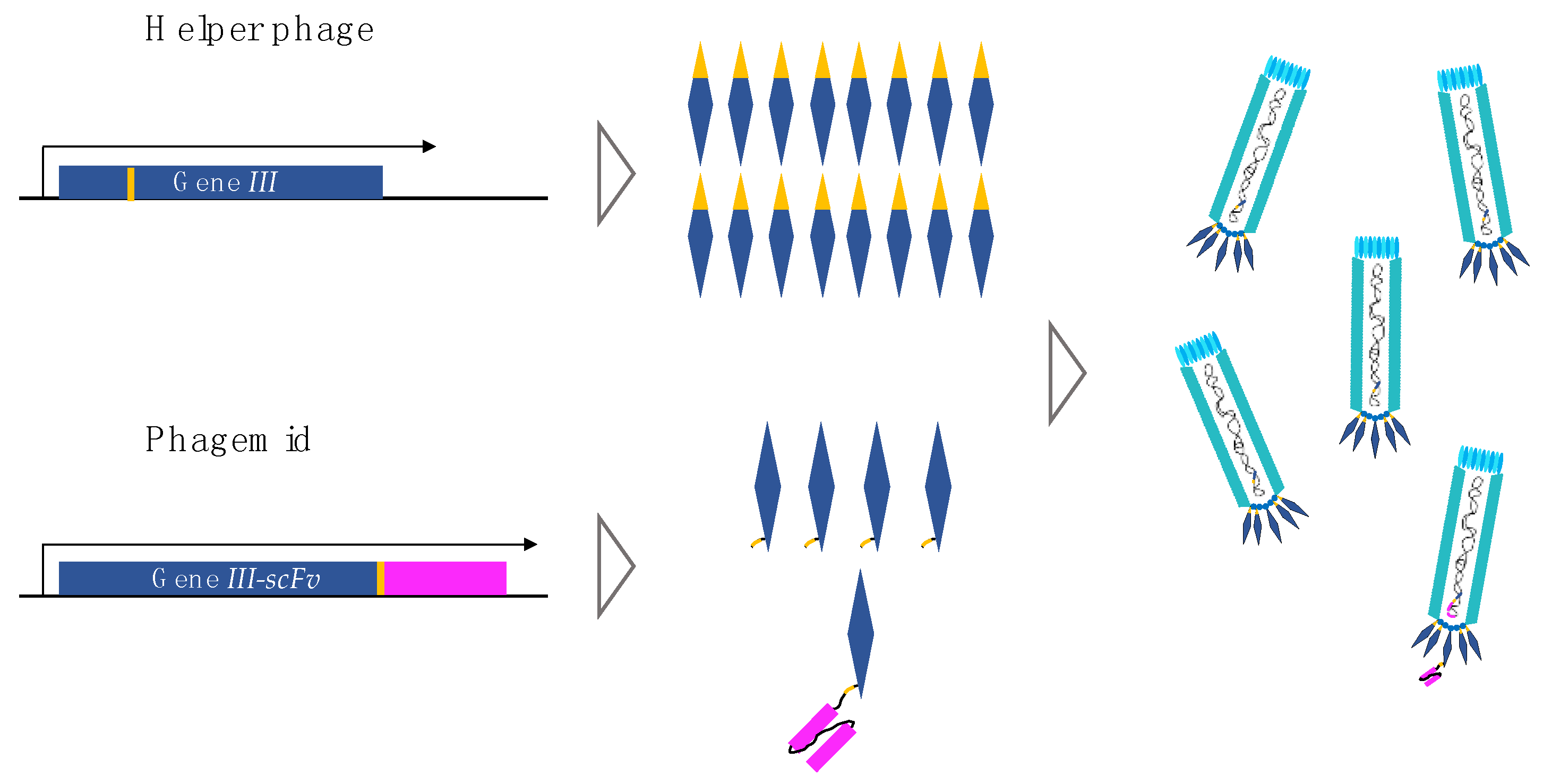
4. Phage and Phagemid Libraries
5. Phagemid Libraries Are Amplified Using Helper Phages
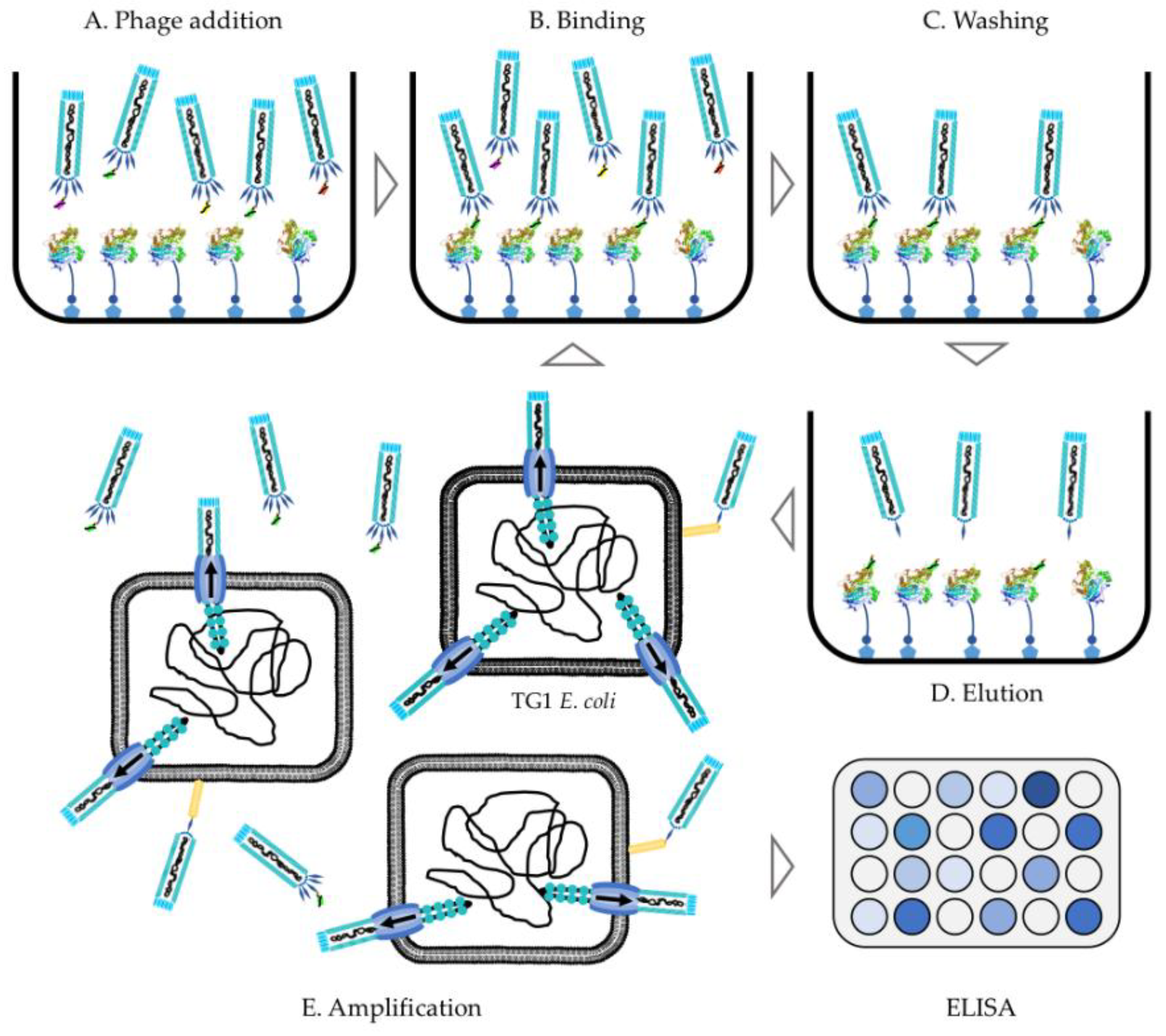
6. Performing a Phage Display Selection Experiment
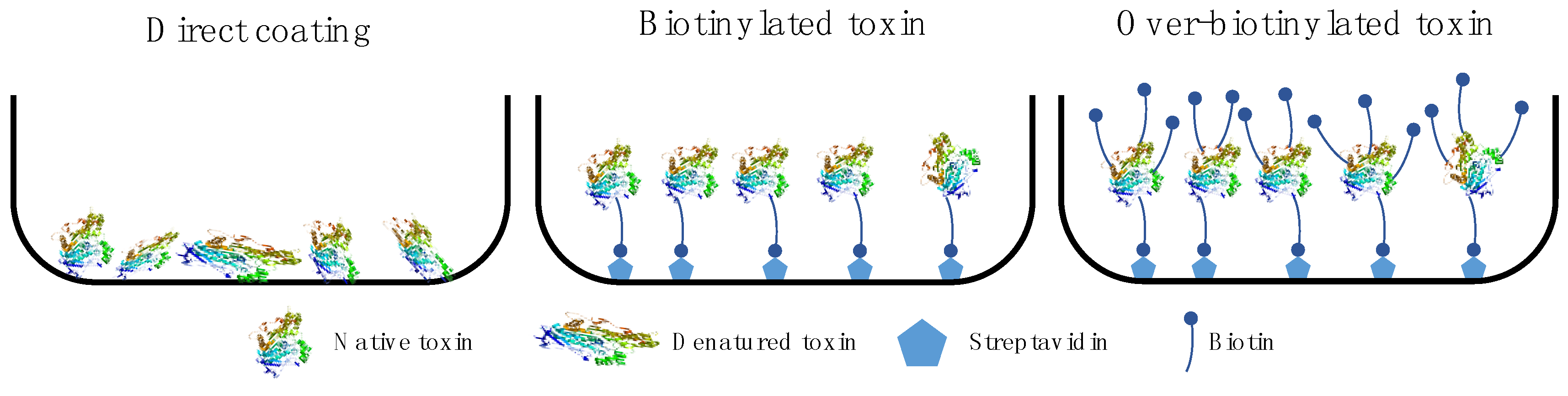
7. Antigen Quality and Presentation
8. Naïve versus Immunized Phage Display Antibody Libraries
9. Selected Examples of the Use of Antibody Phage Display Selection within Toxinology
10. Closing Remarks
Acknowledgments
Conflicts of Interest
References
- Chippaux, J.-P. Snakebite envenomation turns again into a neglected tropical disease! J. Venom. Anim. Toxins Trop. Dis. 2017, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laustsen, A.H.; Engmark, M.; Milbo, C.; Johannesen, J.; Lomonte, B.; Gutiérrez, J.M.; Lohse, B. From Fangs to Pharmacology: The Future of Snakebite Envenoming Therapy. Curr. Pharm. Des. 2016, 22, 5270–5293. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, A.H. Guiding recombinant antivenom development by omics technologies. New Biotechnol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.A.; Cook, D.A.; Renjifo, C.; Casewell, N.R.; Currier, R.B.; Wagstaff, S.C. Research strategies to improve snakebite treatment: Challenges and progress. J. Proteom. 2011, 74, 1768–1780. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, C.; Laustsen, A.H. Recent Advances in Next Generation Snakebite Antivenoms. Trop. Med. Infect. Dis. 2018, 3, 42. [Google Scholar] [CrossRef]
- Laustsen, A.H.; Johansen, K.H.; Engmark, M.; Andersen, M.R. Recombinant snakebite antivenoms: A cost-competitive solution to a neglected tropical disease? PLoS Negl. Trop. Dis. 2017, 11, e0005361. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Meyers, A.J.; McLean, M.D.; Arbabi-Ghahroudi, M.; MacKenzie, R.; Hall, J.C. In Vivo Neutralization of α-Cobratoxin with High-Affinity Llama Single-Domain Antibodies (VHHs) and a VHH-Fc Antibody. PLoS ONE 2013, 8, e69495. [Google Scholar] [CrossRef] [PubMed]
- Julve Parreño, J.M.; Huet, E.; Fernández-Del-Carmen, A.; Segura, A.; Venturi, M.; Gandía, A.; Pan, W.-S.; Albaladejo, I.; Forment, J.; Pla, D.; et al. A synthetic biology approach for consistent production of plant-made recombinant polyclonal antibodies against snake venom toxins. Plant Biotechnol. J. 2018, 16, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, A.H.; Johansen, K.H.; Engmark, M.; Andersen, M.R. Snakebites: Costing recombinant antivenoms. Nature 2016, 538, 41. [Google Scholar] [CrossRef] [PubMed]
- Roncolato, E.C.; Campos, L.B.; Pessenda, G.; Costa e Silva, L.; Furtado, G.P.; Barbosa, J.E. Phage display as a novel promising antivenom therapy: A review. Toxicon 2015, 93, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Bazan, J.; Całkosiński, I.; Gamian, A. Phage display—A powerful technique for immunotherapy. Hum. Vaccines Immunother. 2012, 8, 1817–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laustsen, A.H.; Solà, M.; Jappe, E.C.; Oscoz, S.; Lauridsen, L.P.; Engmark, M. Biotechnological Trends in Spider and Scorpion Antivenom Development. Toxins 2016, 8, 226. [Google Scholar] [CrossRef] [PubMed]
- Hofschneider, P.H. Untersuchungen über „kleine “E. coli K 12 Bakteriophagen. Z. Für Naturforschung B 2014, 18, 203–210. [Google Scholar] [CrossRef]
- Rasched, I.; Oberer, E. Ff coliphages: Structural and functional relationships. Microbiol. Rev. 1986, 50, 401–427. [Google Scholar] [PubMed]
- O’Callaghan, R.; Bradley, R.; Paranchych, W. The effect of M13 phage infection upon the F pili of E. coli. Virology 1973, 54, 220–229. [Google Scholar] [CrossRef]
- van Wezenbeek, P.M.; Hulsebos, T.J.; Schoenmakers, J.G. Nucleotide sequence of the filamentous bacteriophage M13 DNA genome: Comparison with phage fd. Gene. 1980, 11, 129–148. [Google Scholar] [CrossRef]
- Berkowitz, S.A.; Day, L.A. Mass, length, composition and structure of the filamentous bacterial virus fd. J. Mol. Biol. 1976, 102, 531–547. [Google Scholar] [CrossRef]
- Jacobson, A. Role of F Pili in the Penetration of Bacteriophage fl. J. Virol. 1972, 10, 835–843. [Google Scholar] [PubMed]
- Riechmann, L.; Holliger, P. The C-terminal domain of TolA is the coreceptor for filamentous phage infection of E. coli. Cell 1997, 90, 351–360. [Google Scholar] [CrossRef]
- Click, E.M.; Webster, R.E. Filamentous phage infection: Required interactions with the TolA protein. J. Bacteriol. 1997, 179, 6464–6471. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.; Maddera, L.; Harris, R.L.; Silverman, P.M. F-pili dynamics by live-cell imaging. Proc. Natl. Acad. Sci. USA. 2008, 105, 17978–17981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clackson, T.; Lowman, H. Phage Display: A Practical Approach; Oxford University Press: Oxford, UK, 2004. [Google Scholar]
- Branston, S. An Investigation of the Properties of Bacteriophage M13 and the Implications for Its Large-Scale Bioprocessing. Ph.D. Thesis, University College London, London, UK, 2009. [Google Scholar]
- Russel, M.; Whirlow, H.; Sun, T.P.; Webster, R.E. Low-frequency infection of F- bacteria by transducing particles of filamentous bacteriophages. J. Bacteriol. 1988, 170, 5312–5316. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.E. The tol gene products and the import of macromolecules into Escherichia coli. Mol. Microbiol. 1991, 5, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Suggs, S.V. Replication of bacteriophage M13. J. Mol. Biol. 1977, 110, 147–163. [Google Scholar] [CrossRef]
- Weigel, C. Bacteriophage replication modules. FEMS Microbiol. Rev. 2006, 30, 321–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asano, S.; Higashitani, A.; Horiuchi, K. Filamentous phage replication initiator protein gpII forms a covalent complex with the 5′ end of the nick it introduced. Nucleic Acids Res. 1999, 27, 1882–1889. [Google Scholar] [CrossRef] [PubMed]
- Stassen, A.P. Single-stranded DNA binding protein encoded by the filamentous bacteriophage M13: Structural and functional characteristics. Mol. Biol. Rep. 1994, 20, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Russel, M.; Model, P. Genetic analysis of the filamentous bacteriophage packaging signal and of the proteins that interact with it. J. Virol. 1989, 63, 3284–3295. [Google Scholar] [PubMed]
- Russel, M.; Model, P. The role of thioredoxin in filamentous phage assembly. Construction, isolation, and characterization of mutant thioredoxins. J. Biol. Chem. 1986, 261, 14997–15005. [Google Scholar] [PubMed]
- Rakonjac, J. Filamentous phage are released from the bacterial membrane by a two-step mechanism involving a short C-terminal fragment of pIII. J. Mol. Biol. 1999, 289, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Rakonjac, J.; Model, P. Roles of pIII in filamentous phage assembly. J. Mol. Biol. 1998, 282, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Rakonjac, J.; Jovanovic, G.; Model, P. Filamentous phage infection-mediated gene expression: Construction and propagation of the gIII deletion mutant helper phage R408d3. Gene. 1997, 198, 99–103. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science. 1985, 228, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature. 1990, 348, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Dooley, H. Selection and characterization of naturally occurring single-domain (IgNAR) antibody fragments from immunized sharks by phage display. Mol. Immunol. 2003, 40, 25–33. [Google Scholar] [CrossRef]
- Sidhu, S.S. Engineering M13 for phage display. Biomol. Eng. 2001, 18, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Mandrup, O.A.; Friis, N.A.; Lykkemark, S.; Just, J.; Kristensen, P. A Novel Heavy Domain Antibody Library with Functionally Optimized Complementarity Determining Regions. PLoS ONE. 2013, 8, e76834. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.; Buxbaum, S.; Link, J.; Smith, R.; Venti, C.; Darsley, M. Determination of binding constants of diabodies directed against prostate-specific antigen using electrochemiluminescence-based immunoassays. J. Mol. Recognit. 1996, 9, 456–461. [Google Scholar] [CrossRef]
- Schofield, D.J.; Pope, A.R.; Clementel, V.; Buckell, J.; Chapple, S.D.; Clarke, K.F.; Conquer, J.S.; Crofts, A.M.; Crowther, S.R.; Dyson, M.R.; et al. Application of phage display to high throughput antibody generation and characterization. Genome Biol. 2007, 8, R254. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboom, H.R.; de Bruïne, A.P.; Hufton, S.E.; Hoet, R.M.; Arends, J.-W.; Roovers, R.C. Antibody phage display technology and its applications. Immunotechnology. 1998, 4, 1–20. [Google Scholar] [CrossRef]
- O’Connell, D.; Becerril, B.; Roy-Burman, A.; Daws, M.; Marks, J.D. Phage versus phagemid libraries for generation of human monoclonal antibodies. J. Mol. Biol. 2002, 321, 49–56. [Google Scholar] [CrossRef]
- Rondot, S.; Koch, J.; Breitling, F.; Dübel, S. A helper phage to improve single-chain antibody presentation in phage display. Nat. Biotechnol. 2001, 19, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Vieira, J.; Messing, J. Production of single-stranded plasmid DNA. In Methods in Enzymology; Recombinant DNA Part D; Academic Press: Cambridge, MA, USA, 1987; Volume 153, pp. 3–11. [Google Scholar]
- Kristensen, P.; Winter, G. Proteolytic selection for protein folding using filamentous bacteriophages. Fold. Des. 1998, 3, 321–328. [Google Scholar] [CrossRef]
- Lou, J. Affinity Maturation by Chain Shuffling and Site Directed Mutagenesis. In Antibody Engineering, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 1, pp. 377–396. [Google Scholar]
- Laustsen, A.H.; Lauridsen, L.P.; Lomonte, B.; Andersen, M.R.; Lohse, B. Pitfalls to avoid when using phage display for snake toxins. Toxicon. 2017, 126, 79–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laustsen, A.H. Recombinant Antivenoms, 1st ed.; University of Copenhagen: Copenhagen, Denmark, 2016; ISBN 978-87-93086-61-6. [Google Scholar]
- Stewart, C.S.; MacKenzie, C.R.; Hall, J.C. Isolation, characterization and pentamerization of alpha-cobrotoxin specific single-domain antibodies from a naïve phage display library: Preliminary findings for antivenom development. Toxicon. 2007, 49, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Marks, J.D. By-passing immunization. J. Mol. Biol. 1991, 222, 581–597. [Google Scholar] [CrossRef]
- Hawkins, R.E.; Russell, S.J.; Winter, G. Selection of phage antibodies by binding affinity: Mimicking affinity maturation. J. Mol. Biol. 1992, 226, 889–896. [Google Scholar] [CrossRef]
- Lowe, D.; Wilkinson, T.; Vaughan, T.J. Affinity Maturation Approaches for Antibody Lead Optimization. In Antibody Drug Discovery; Molecular Medicine and Medicinal Chemistry; Imperial College Press: London, UK, 2011; Volume 4, pp. 85–119. ISBN 978-1-84816-628-8. [Google Scholar]
- Meng, J.; John, T.R.; Kaiser, I.I. Specificity and binding affinity of an anti-crotoxin combinatorial antibody selected from a phage-displayed library. Biochem. Pharmacol. 1995, 50, 1969–1977. [Google Scholar] [CrossRef] [PubMed]
- Kulkeaw, K.; Sakolvaree, Y.; Srimanote, P.; Tongtawe, P.; Maneewatch, S.; Sookrung, N.; Tungtrongchitr, A.; Tapchaisri, P.; Kurazono, H.; Chaicumpa, W. Human monoclonal ScFv neutralize lethal Thai cobra, Naja kaouthia, neurotoxin. J. Proteom. 2009, 72, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rodríguez, E.R.; Olamendi-Portugal, T.; Serrano-Posada, H.; Arredondo-López, J.N.; Gómez-Ramírez, I.; Fernández-Taboada, G.; Possani, L.D.; Anguiano-Vega, G.A.; Riaño-Umbarila, L.; Becerril, B. Broadening the neutralizing capacity of a family of antibody fragments against different toxins from Mexican scorpions. Toxicon. 2016, 119, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Pucca, M.B.; Zoccal, K.F.; Roncolato, E.C.; Bertolini, T.B.; Campos, L.B.; Cologna, C.T.; Faccioli, L.H.; Arantes, E.C.; Barbosa, J.E. Serrumab: A human monoclonal antibody that counters the biochemical and immunological effects of Tityus serrulatus venom. J. Immunotoxicol. 2012, 9, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Pucca, M.B.; Cerni, F.A.; Peigneur, S.; Arantes, E.C.; Tytgat, J.; Barbosa, J.E. Serrumab: A novel human single chain-fragment antibody with multiple scorpion toxin-neutralizing capacities. J. Immunotoxicol. 2014, 11, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Pessenda, G.; Silva, L.C.; Campos, L.B.; Pacello, E.M.; Pucca, M.B.; Martinez, E.Z.; Barbosa, J.E. Human scFv antibodies (Afribumabs) against Africanized bee venom: Advances in melittin recognition. Toxicon. 2016, 112, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.-P.; Chen, I.-C.; Yu, C.-M.; Peng, H.-P.; Jian, J.-W.; Ma, S.-H.; Lee, Y.-C.; Jan, J.-T.; Yang, A.-S. Discovering neutralizing antibodies targeting the stem epitope of H1N1 influenza hemagglutinin with synthetic phage-displayed antibody libraries. Sci. Rep. 2015, 5, 15053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeld, R.; Alcalay, R.; Mechaly, A.; Lapidoth, G.; Epstein, E.; Kronman, C.; J Fleishman, S.; Mazor, O. Improved antibody-based ricin neutralization by affinity maturation is correlated with slower off-rate values. Protein Eng. Des. Sel. 2017, 30, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Bakker, A.B.H.; Marissen, W.E.; Kramer, R.A.; Rice, A.B.; Weldon, W.C.; Niezgoda, M.; Hanlon, C.A.; Thijsse, S.; Backus, H.H.J.; de Kruif, J.; et al. Novel human monoclonal antibody combination effectively neutralizing natural rabies virus variants and individual in vitro escape mutants. J. Virol. 2005, 79, 9062–9068. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, A.H.; María Gutiérrez, J.; Knudsen, C.; Johansen, K.H.; Bermúdez-Méndez, E.; Cerni, F.A.; Jürgensen, J.A.; Ledsgaard, L.; Martos-Esteban, A.; Øhlenschlæger, M.; et al. Pros and cons of different therapeutic antibody formats for recombinant antivenom development. Toxicon 2018, 146, 151–175. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Protein Name (Abbreviation) | Size (kDa) | Function |
|---|---|---|---|
| I | Gene 1 protein (G1P) | 39.6 | Assembly |
| Gene 11 protein (G11P) | 12.4 | Assembly | |
| II | Replication-associated protein (G2P) | 46.2 | Replication |
| Gene 10 protein (G10P) | 12.7 | Replication | |
| III | Attachment protein (G3P) | 44.7 | Coat protein Adsorption and extrusion |
| IV | Virion export protein (G4P) | 45.9 | Assembly and extrusion |
| V | DNA-binding protein (G5P) | 9.7 | Replication |
| VI | Head virion protein (G6P) | 12.4 | Coat protein Infection and budding |
| VII | Tail virion protein (G7P) | 3.6 | Coat protein Assembly and budding |
| VIII | Capsid protein (G8P) | 7.6 | Coat protein |
| IX | Tail virion protein (G9P) | 3.7 | Coat protein Assembly and budding |
| Genetic Element | Phagemid | Helper Phage |
|---|---|---|
| Gene I | X | |
| Gene II | X | |
| Gene III | X | |
| Gene IV | X | |
| Gene V | X | |
| Gene VI | X | |
| Gene VII | X | |
| Gene VIII | X | |
| Gene IX | X | |
| Gene III-scFv | X | |
| Antibiotic resistance gene | X | X |
| Origin of replication | X | |
| Origin of replication (inefficient) | X |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ledsgaard, L.; Kilstrup, M.; Karatt-Vellatt, A.; McCafferty, J.; Laustsen, A.H. Basics of Antibody Phage Display Technology. Toxins 2018, 10, 236. https://doi.org/10.3390/toxins10060236
Ledsgaard L, Kilstrup M, Karatt-Vellatt A, McCafferty J, Laustsen AH. Basics of Antibody Phage Display Technology. Toxins. 2018; 10(6):236. https://doi.org/10.3390/toxins10060236
Chicago/Turabian StyleLedsgaard, Line, Mogens Kilstrup, Aneesh Karatt-Vellatt, John McCafferty, and Andreas H. Laustsen. 2018. "Basics of Antibody Phage Display Technology" Toxins 10, no. 6: 236. https://doi.org/10.3390/toxins10060236
APA StyleLedsgaard, L., Kilstrup, M., Karatt-Vellatt, A., McCafferty, J., & Laustsen, A. H. (2018). Basics of Antibody Phage Display Technology. Toxins, 10(6), 236. https://doi.org/10.3390/toxins10060236