LC-MS/MS and LC-UV Determination of Moniliformin by Adding Lanthanide Ions to the Mobile Phase


Abstract
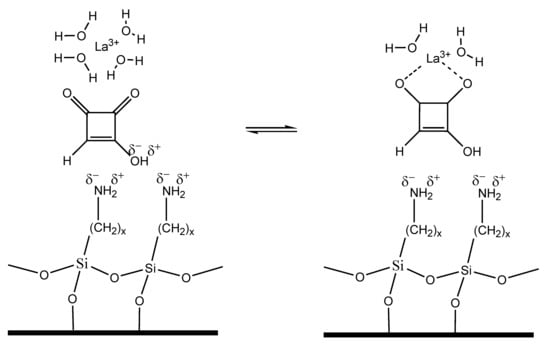
:
1. Introduction
2. Results and Discussion
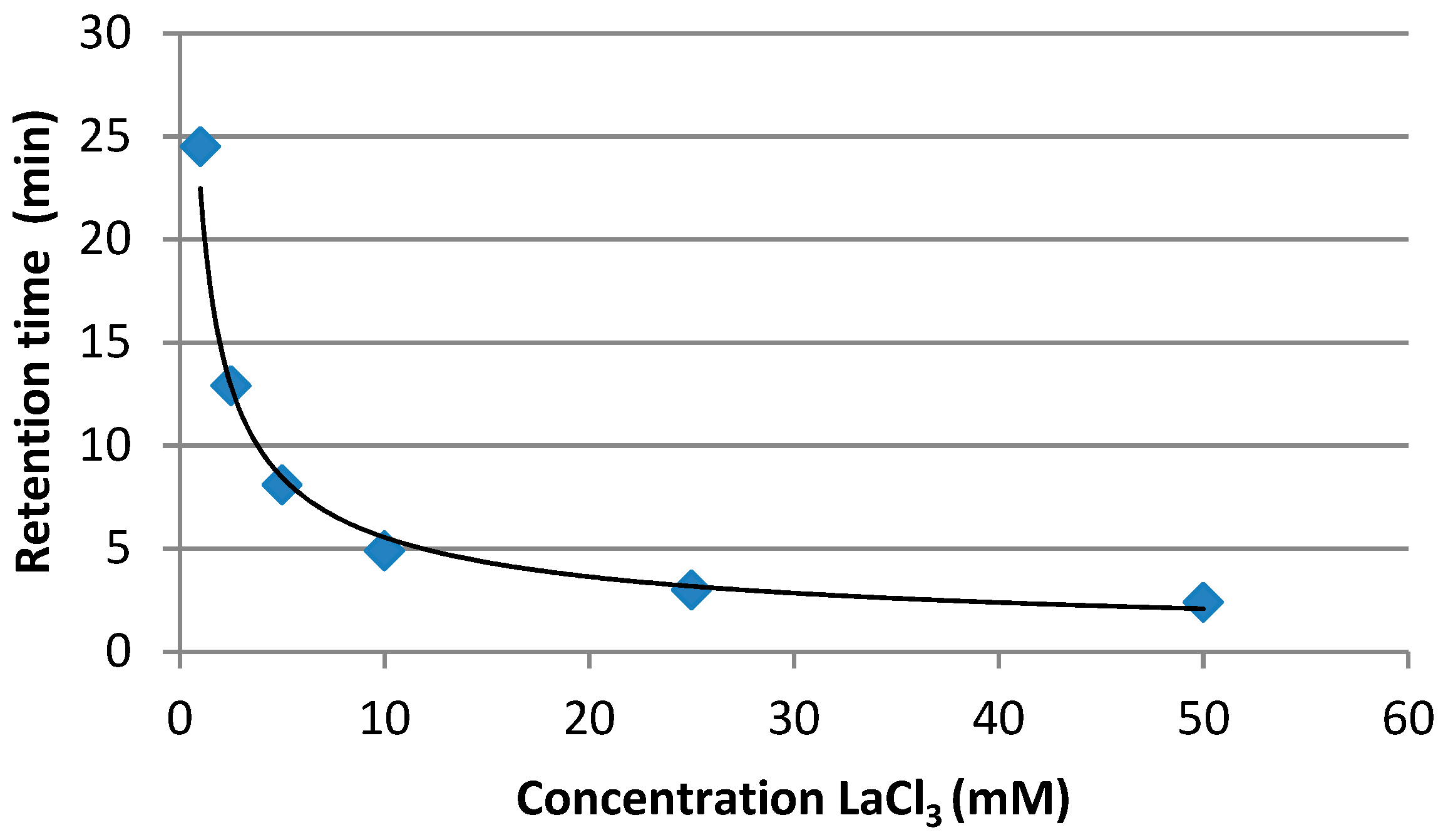
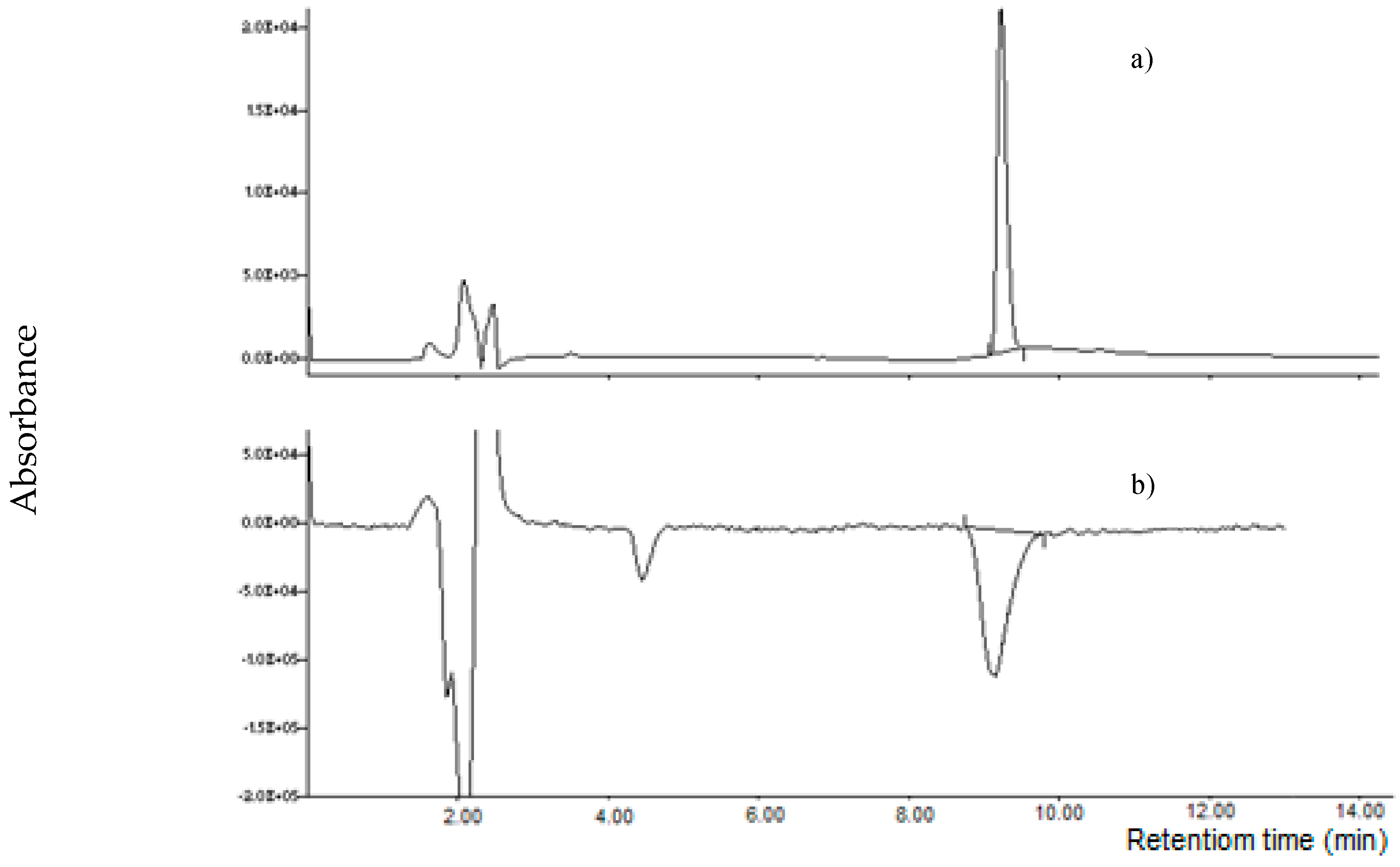
2.1. Development of the Chromatographic Method
2.2. Development of an Extraction and Purification Method for Cereal Samples
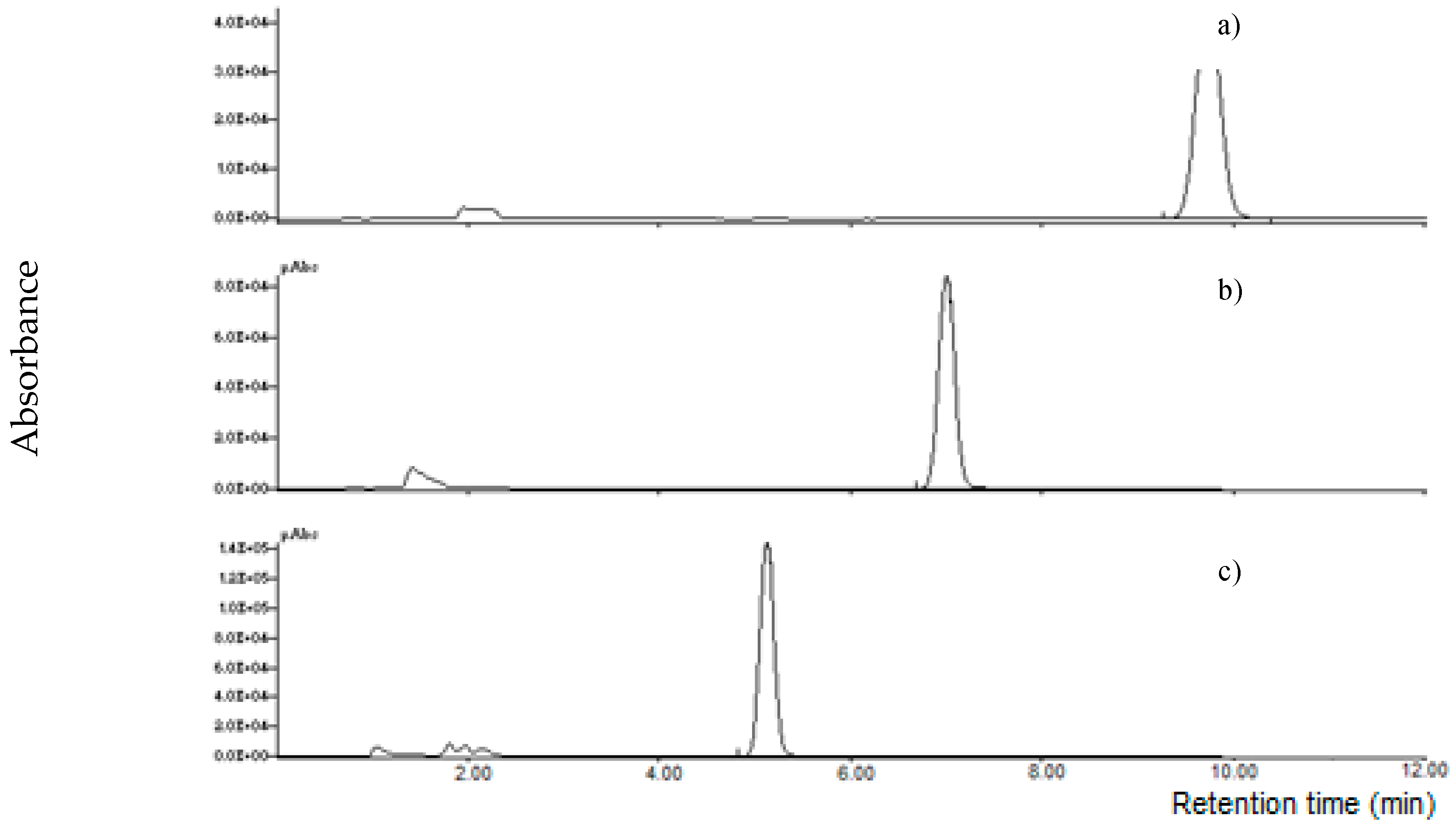
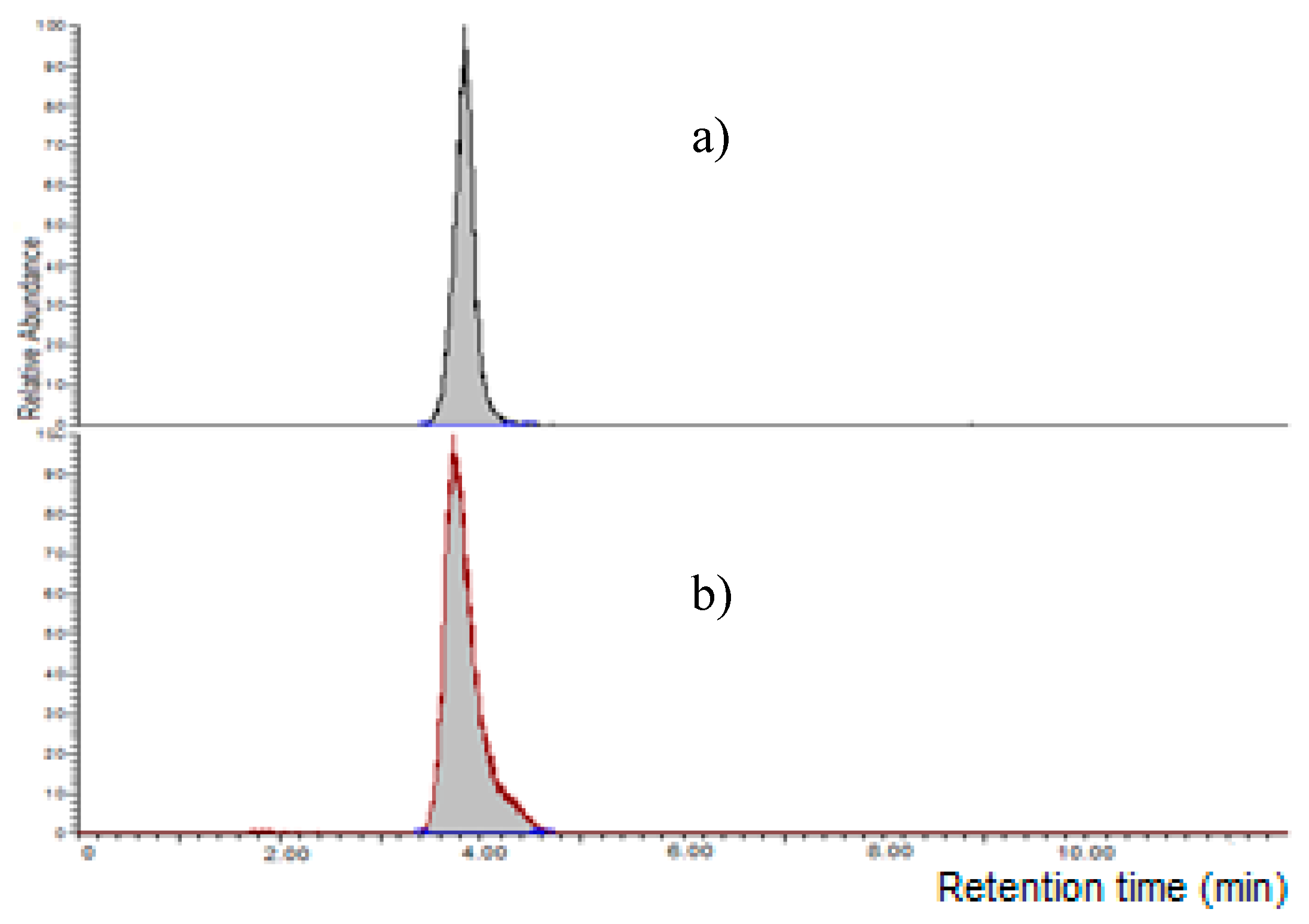
2.3. Performances of the Method
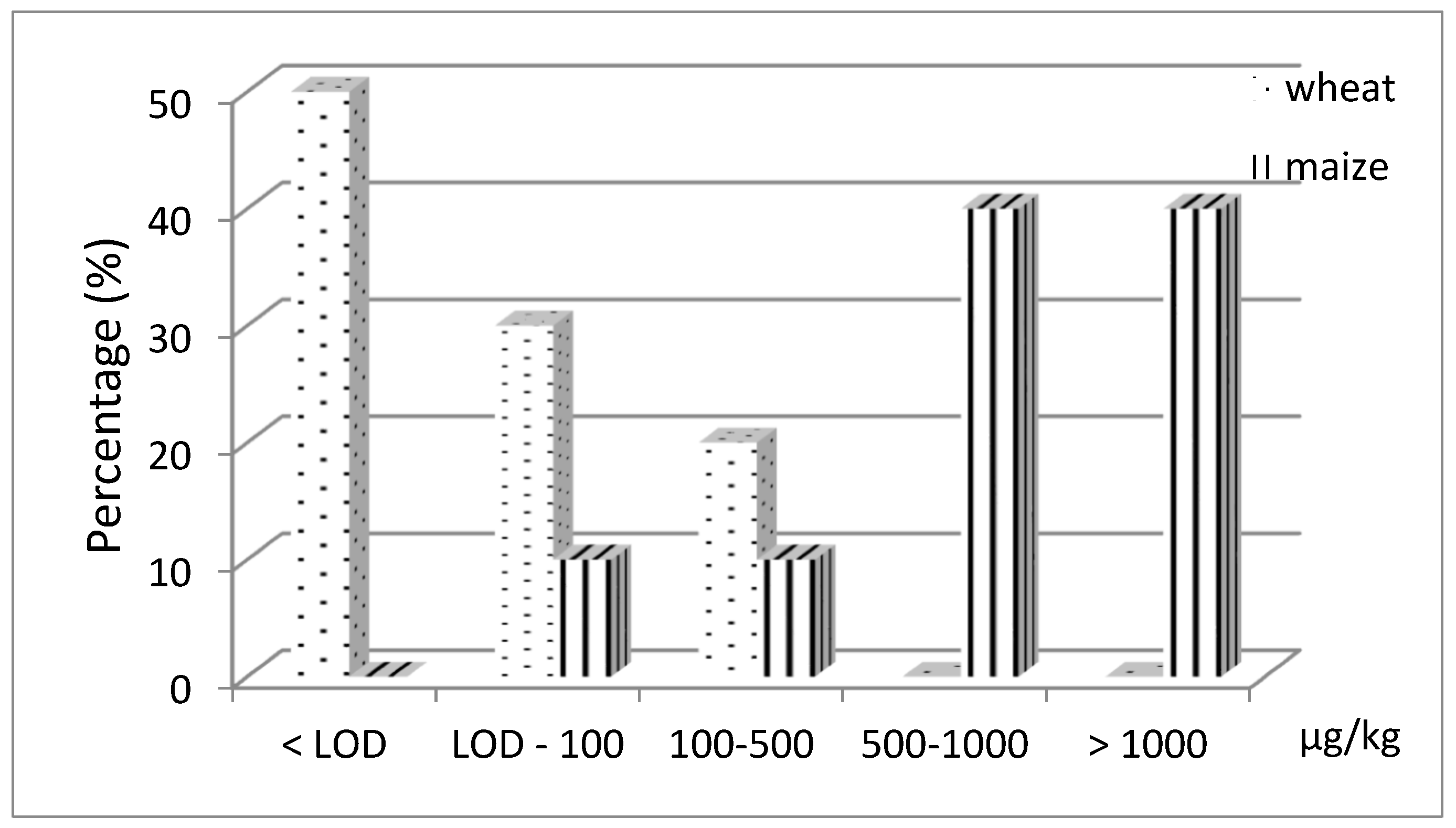
2.4. MON Occurrence in Cereal Samples
3. Materials and Methods
3.1. Reagents and Standards
3.2. LC-MS/MS Analysis for Moniliformin Determination
3.3. HPLC-UV Analysis for Moniliformin Determination
3.4. Method Validation
3.5. Real Sample Collection and Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Uhlig, S.; Torp, M.; Jarp, J.; Parich, A.; Gutleb, A.C.; Krska, R. Moniliformin in Norwegian grain. Food Addit. Contam. 2004, 21, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Steyn, M.; Thiel, P.G.; Van Schalkwyk, G.C. Isolation and purification of moniliformin. J. Assoc. Off. Anal. Chem. 1978, 61, 578–580. [Google Scholar] [PubMed]
- Kriek, N.; Marasas, W.; Steyn, P.; Van Rensburg, S.; Steyn, M. Toxicity of a moniliformin-producing strain of fusarium moniliforme var. subglutinans isolated from maize. Food Cosmet. Toxicol. 1977, 15, 579–587. [Google Scholar] [CrossRef]
- Jestoi, M. Emerging Fusarium-Mycotoxins Fusaproliferin, Beauvericin, Enniatins, and Moniliformin—A Review. Crit. Rev. Food Sci. Nutr. 2008, 48, 21–49. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel Contam. Risks to human and animal health related to the presence of moniliformin in food and feed. EFSA J. 2018, 16, 5082. [Google Scholar]
- Rokka, M.; Yli-Mattila, T.; Parikka, P.; Rizzo, A.; Peltonen, K.; Jestoi, M. Presence and concentrations of theFusarium-related mycotoxins beauvericin, enniatins and moniliformin in Finnish grain samples. Food Addit. Contam. 2004, 21, 794–802. [Google Scholar]
- Van Asselt, E.; Azambuja, W.; Moretti, A.; Kastelein, P.; De Rijk, T.; Stratakou, I.; Van Der Fels-Klerx, H. A Dutch field survey on fungal infection and mycotoxin concentrations in maize. Food Addit. Contam. Part A 2012, 29, 1556–1565. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, V.; Blandino, M.; Negre, M.; Reyneri, A.; Vanara, F. Moniliformin analysis in maize samples from North-West Italy using multifunctional clean-up columns and the LC-MS/MS detection method. Food Addit. Contam. 2013, 30, 876–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Bargen, K.W.; Lohrey, L.; Cramer, B.; Humpf, H.-U. Analysis of the Fusarium mycotoxin moniliformin in cereals samples using 13C2-moniliformin and High- Resolution Mass Spectrometry. J. Agric. Food Chem. 2012, 60, 3586–3591. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.; Van Dam, R.; Spanjer, M.; De Stoppelaar, J.; Mol, H.; De Nijs, M.; López, P. Survey of moniliformin in wheat- and corn-based products using a straightforward analytical method. Mycotoxin Res. 2017, 33, 333–341. [Google Scholar] [CrossRef]
- Barthel, J.; Rapp, M.; Holtmannspötter, H.; Gottschalk, C. A rapid LC-MS/MS method for the determination of moniliformin and occurrence of this mycotoxin in maize products from the Bavarian market. Mycotoxin Res. 2018, 349, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, M.J.; Gilbert, J. Method for the analysis in maize of the fusarium mycotoxin moniliformin employing ion-pairing extraction and high-performance liquid chromatography. J. Chromatogr. A 1986, 358, 415–422. [Google Scholar] [CrossRef]
- Lim, C.W.; Lai, K.Y.; Yeo, J.F.; Tai, S.H.; Chan, S.H. Quantitative assessment of moniliformin in cereals via alternative precipitation pathways, aided by LC-LIT-MS and LC-Q-TOF-MS. Food Chem. 2015, 174, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Hallas-Møller, M.; Frisvad, J.C.; Nielsen, K.F. Production of the Fusarium Mycotoxin Moniliformin by Penicillium melanoconidium. J. Agric. Food Chem. 2016, 64, 4505–4510. [Google Scholar] [CrossRef] [PubMed]
- Binnemans, K. Rare-earth beta-diketonates. In Handbook of the Physics and Chemistry of Rare Earths; Gschneidner, K.A., Jr., Bünzli, J.-C.G., Pecharsky, V.K., Eds.; North Holland: Amsterdam, Holland, 2005; Volume 35, pp. 107–272. [Google Scholar]
- Maragos, C.M. Complexation of the Mycotoxin Cyclopiazonic Acid with Lanthanides Yields Luminescent Products. Toxins 2018, 10, 285. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, B.; Fente, C.; Franco, C.; Cepeda, A.; Prognon, P.; Mahuzier, G. Simultaneous high-performance liquid chromatographic determination of ochratoxin A and citrinin in cheese by time-resolved luminescence using terbium. J. Chromatogr. A 1996, 727, 185–193. [Google Scholar] [CrossRef]
- Armelao, L.; Quici, S.; Barigelletti, F.; Accorsi, G.; Bottaro, G.; Cavazzini, M.; Tondello, E. Design of luminescent lanthanide complexes: From molecules to highly efficient photo-emitting materials. Coord. Chem. Rev. 2010, 254, 487–505. [Google Scholar] [CrossRef]
- Rocha, J.; Carlos, L.D.; Paz, F.A.; Ananias, D. Luminescent multifunctional lanthanides-based metal-organic frameworks. Chem. Soc. Rev. 2011, 40, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Davankov, V.A.; Navratil, J.D.; Walton, H.F. Ligand Exchange Chromatography; CRC Press: Boca Raton, FL, USA, 1988. [Google Scholar]
- Commission Decision of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results 2002/657/EC. Off. J. Eur. Communities 2002, L221, 8–36.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HPLC-UV | LC-MS/MS | |
|---|---|---|
| Matrix effect (at 800 µg kg−1) * | / | 5% (wheat) |
| 8% (maize) | ||
| Calibration range * | 160–8000 µg kg−1 | 20–2000 µg kg−1 |
| LOD * | 80 µg kg−1 | 10 µg kg−1 |
| LOQ * | 200 µg kg−1 | 25 µg kg−1 |
| Average recovery | ||
| Wheat | 88.2% ± 5.5% | 97.1% ± 4.3% |
| Maize | 84.8% ± 5.4% | 96.4% ± 5.1% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertuzzi, T.; Rastelli, S.; Mulazzi, A.; Pietri, A. LC-MS/MS and LC-UV Determination of Moniliformin by Adding Lanthanide Ions to the Mobile Phase. Toxins 2019, 11, 570. https://doi.org/10.3390/toxins11100570
Bertuzzi T, Rastelli S, Mulazzi A, Pietri A. LC-MS/MS and LC-UV Determination of Moniliformin by Adding Lanthanide Ions to the Mobile Phase. Toxins. 2019; 11(10):570. https://doi.org/10.3390/toxins11100570
Chicago/Turabian StyleBertuzzi, Terenzio, Silvia Rastelli, Annalisa Mulazzi, and Amedeo Pietri. 2019. "LC-MS/MS and LC-UV Determination of Moniliformin by Adding Lanthanide Ions to the Mobile Phase" Toxins 11, no. 10: 570. https://doi.org/10.3390/toxins11100570
APA StyleBertuzzi, T., Rastelli, S., Mulazzi, A., & Pietri, A. (2019). LC-MS/MS and LC-UV Determination of Moniliformin by Adding Lanthanide Ions to the Mobile Phase. Toxins, 11(10), 570. https://doi.org/10.3390/toxins11100570