Development of an UPLC-MS/MS Method for the Analysis of Mycotoxins in Rumen Fluid with and without Maize Silage Emphasizes the Importance of Using Matrix-Matched Calibration

 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Mycotoxin Selection
2.2. UPLC-MS/MS Method
2.3. Sample Preparation and Extraction
2.4. Method Validation with Rumen Fluid–Buffer Mixture as Matrix
2.4.1. Linearity
2.4.2. Accuracy and Precision
2.4.3. Limit of Quantification (LOQ) and Limit of Detection (LOD)
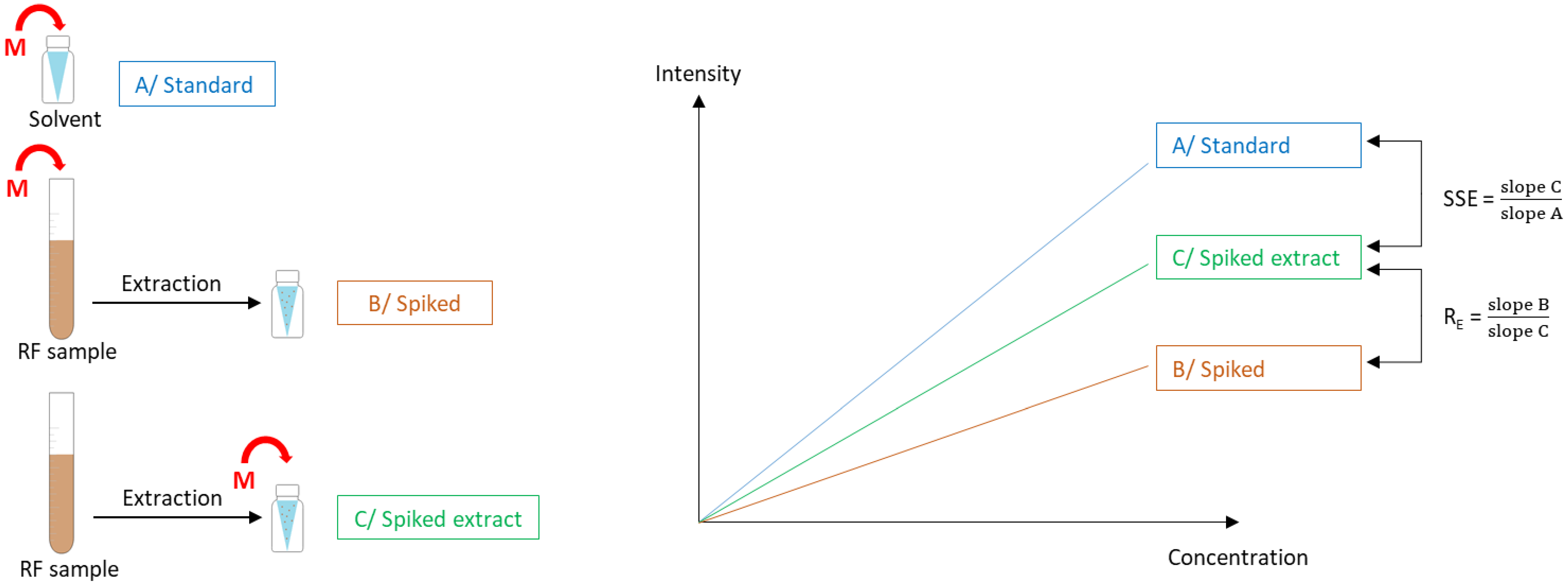
2.4.4. Signal Suppression/Enhancement (SSE) and Extraction Recovery (RE)
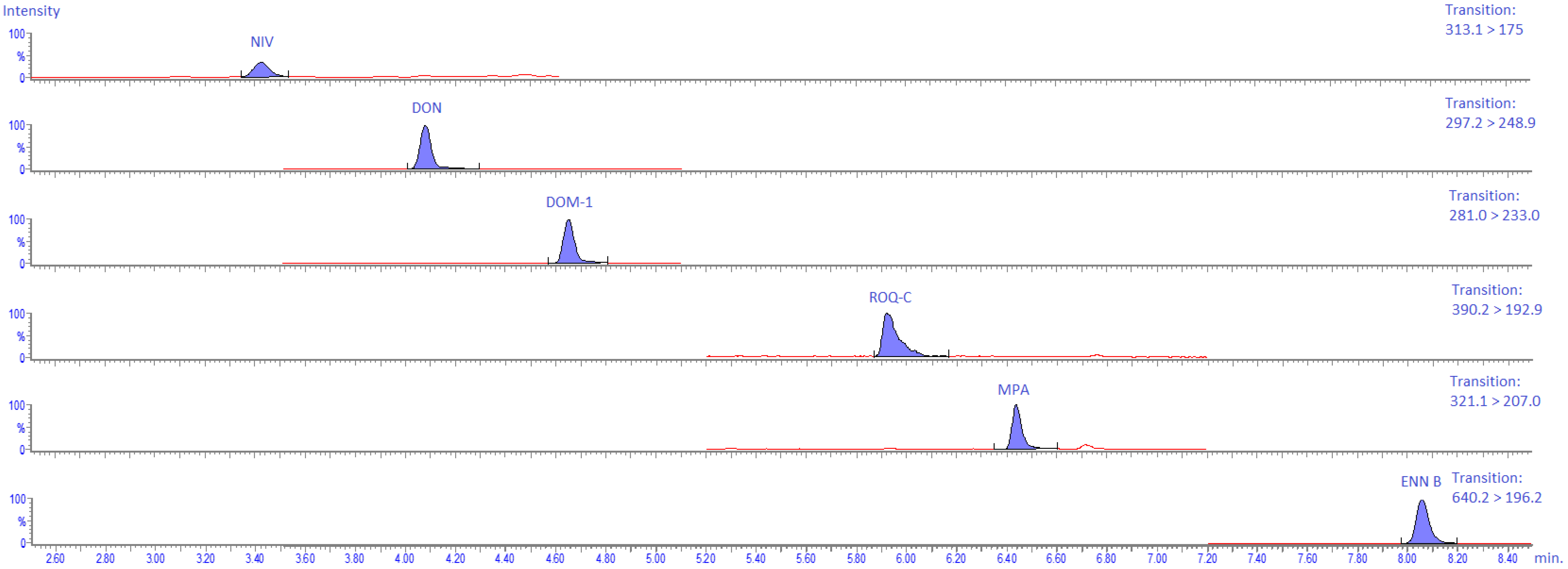
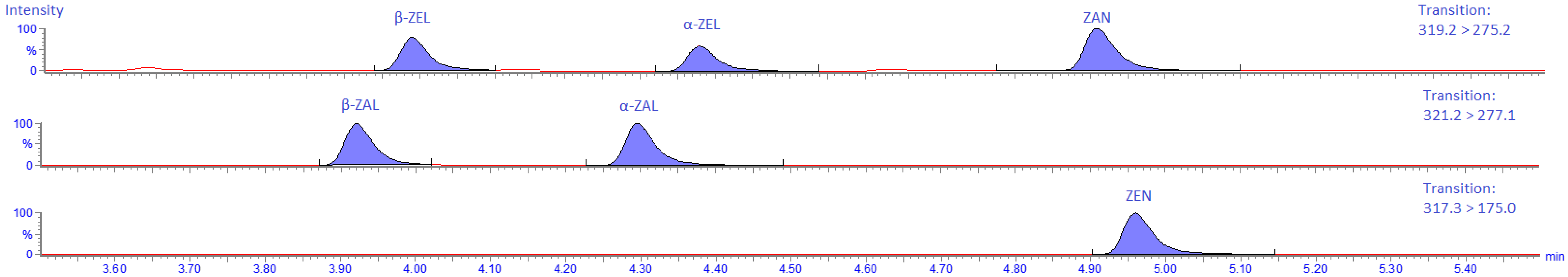
2.4.5. Specificity
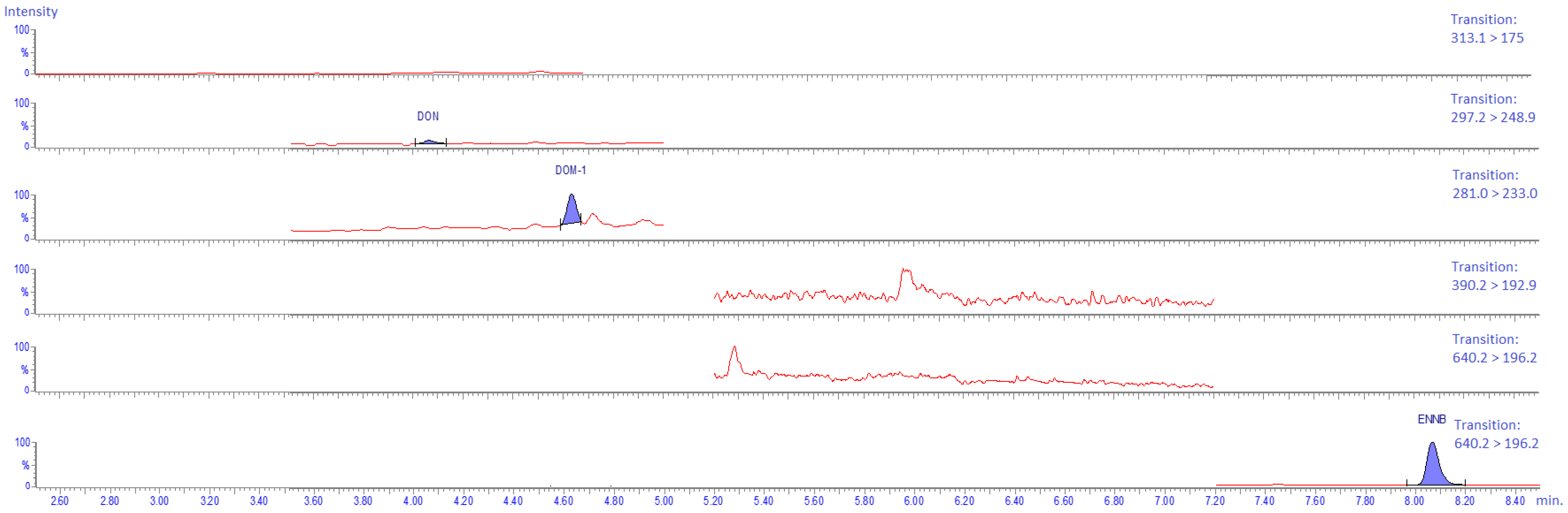
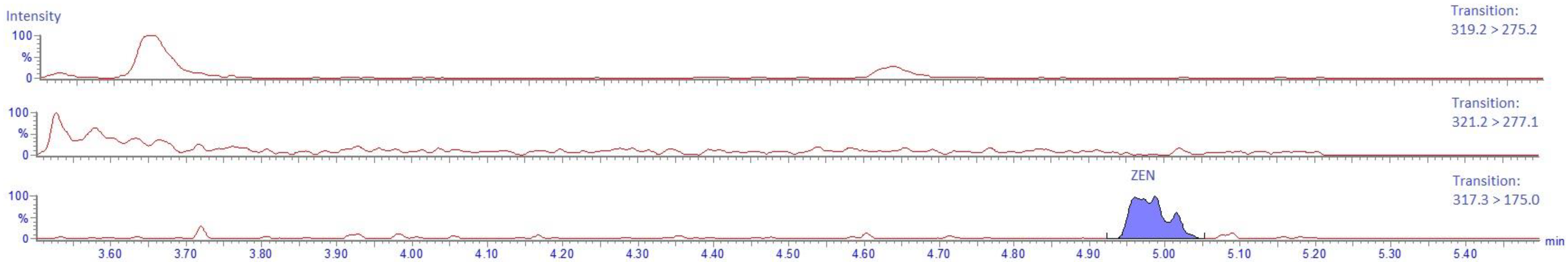
2.4.6. Carry-Over
2.5. Evaluation of the Impact of Maize Silage on the Extraction Recovery (RE) and Signal Suppression/Enhancement (SSE)
2.6. Cross-Validation of the Method with Rumen Fluid and Maize Silage as Matrix
3. Conclusions
4. Materials and Methods
4.1. Mycotoxin Selection
4.2. Rumen Fluid, Maize Silage, Mycotoxins, Chemicals, and Reagents
4.3. Preparation of Standard Solutions and Rumen Fluid–Buffer Mixture
4.4. Preparation of Calibrator, Validation and QC Samples
4.5. Mycotoxin Extraction
4.6. UPLC-MS/MS Analysis
4.7. In-House Method Validation
4.8. Evaluation of the Impact of Maize Silage on the Extraction Recovery (RE) and Signal Suppression/Enhancement (SSE)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fink-Gremmels, J. The role of mycotoxins in the health and performance of dairy cows. Vet. J. 2008, 176, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Upadhaya, S.D.; Park, M.A.; Ha, J.K. Mycotoxins and their biotransformation in the rumen: A review. Asian-Australas. J. Anim. Sci. 2010, 23, 1250–1260. [Google Scholar] [CrossRef]
- Khafipour, E.; Li, S.; Plaizier, J.C.; Krause, D.O. Rumen microbiome composition determined using two nutritional models of subacute ruminal acidosis. Appl. Environ. Microbiol. 2009, 75, 7115–7124. [Google Scholar] [CrossRef] [PubMed]
- Driehuis, F.; Spanjer, M.C.; Scholten, J.M.; Te Giffel, M.C. Occurrence of mycotoxins in maize, grass and wheat silage for dairy cattle in the Netherlands. Food Addit. Contam. Part B 2008, 1, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Zachariasova, M.; Dzuman, Z.; Veprikova, Z.; Hajkova, K.; Jiru, M.; Vaclavikova, M.; Zachariasova, A.; Pospichalova, M.; Florian, M.; Hajslova, J. Occurrence of multiple mycotoxins in European feedingstuffs, assessment of dietary intake by farm animals. Anim. Feed Sci. Technol. 2014, 193, 124–140. [Google Scholar] [CrossRef]
- Valgaeren, B.; Théron, L.; Croubels, S.; Devreese, M.; De Baere, S.; Van Pamel, E.; Daeseleire, E.; De Boevre, M.; De Saeger, S.; Vidal, A.; et al. The role of roughage provision on the absorption and disposition of the mycotoxin deoxynivalenol and its acetylated derivatives in calves: From field observations to toxicokinetics. Arch. Toxicol. 2018, 93, 293–310. [Google Scholar] [CrossRef]
- Tangni, E.K.; Pussemier, L.; Bastiaanse, H.; Haesaert, G.; Foucart, G.; Van Hove, F. Presence of mycophenolic acid, roquefortine C, citrinin and ochratoxin A in maize and grass silages supplied to dairy cattle in Belgium. J. Anim. Sci. Adv. 2013, 3, 598–612. [Google Scholar]
- Driehuis, F.; Spanjer, M.C.; Scholten, J.M.; te Giffel, M.C. Occurrence of mycotoxins in feedstuffs of dairy cows and estimation of total dietary intakes. J. Dairy Sci. 2008, 91, 4261–4271. [Google Scholar] [CrossRef]
- Wambacq, E.; Vanhoutte, I.; Audenaert, K.; De Gelder, L.; Haesaert, G. Occurrence, prevention and remediation of toxigenic fungi and mycotoxins in silage: A review. J. Sci. Food Agric. 2016, 96, 2284–2302. [Google Scholar] [CrossRef]
- Jin, P.G.; Han, Z.; Cai, Z.X.; Wu, Y.J.; Ren, Y.P. Simultaneous determination of 10 mycotoxins in grain by ultra-high-performance liquid chromatography-tandem mass spectrometry using 13C15-deoxynivalenol as internal standard. Food Addit. Contam. Part A 2010, 27, 1701–1713. [Google Scholar] [CrossRef]
- Ren, Y.; Zhang, Y.; Shao, S.; Cai, Z.; Feng, L.; Pan, H.; Wang, Z. Simultaneous determination of multi-component mycotoxin contaminants in foods and feeds by ultra-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2007, 1143, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Van Pamel, E.; Verbeken, A.; Vlaemynck, G.; De Boever, J.; Daeseleire, E. Ultrahigh-performance liquid chromatographic-tandem mass spectrometric multimycotoxin method for quantitating 26 mycotoxins in maize silage. J. Agric. Food Chem. 2011, 59, 9747–9755. [Google Scholar] [CrossRef] [PubMed]
- Broekaert, N.; Devreese, M.; van Bergen, T.; Schauvliege, S.; De Boevre, M.; De Saeger, S.; Vanhaecke, L.; Berthiller, F.; Michlmayr, H.; Malachová, A.; et al. In vivo contribution of deoxynivalenol-3-β-d-glucoside to deoxynivalenol exposure in broiler chickens and pigs: Oral bioavailability, hydrolysis and toxicokinetics. Arch. Toxicol. 2017, 91, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Huybrechts, B.; Martins, J.C.; Debongnie, P.; Uhlig, S.; Callebaut, A. Fast and sensitive LC-MS/MS method measuring human mycotoxin exposure using biomarkers in urine. Arch. Toxicol. 2015, 89, 1993–2005. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.; Claeys, L.; Mengelers, M.; Vanhoorne, V.; Vervaet, C.; Huybrechts, B.; De Saeger, S.; De Boevre, M. Humans significantly metabolize and excrete the mycotoxin deoxynivalenol and its modified form deoxynivalenol-3-glucoside within 24 h. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Gallo, A.; Masoero, F. In vitro models to evaluate the capacity of different sequestering agents to adsorb aflatoxins. Ital. J. Anim. Sci. 2010, 9, 109–116. [Google Scholar] [CrossRef]
- Niderkorn, V.; Morgavi, D.P.; Pujos, E.; Tissandier, A.; Boudra, H. Screening of fermentative bacteria for their ability to bind and biotransform deoxynivalenol, zearalenone and fumonisins in an in vitro model simulating corn silage. Food Addit. Contam. 2007, 24, 406–415. [Google Scholar] [CrossRef]
- Peltonen, K.D.; El-Nezami, H.S.; Salminen, S.J.; Ahokas, J.T. Binding of aflatoxin B1 by probiotic bacteria. J. Sci. Food Agric. 2000, 80, 1942–1945. [Google Scholar] [CrossRef]
- Oatley, J.T.; Rarick, M.D.; Ji, G.E.; Linz, J.E. Binding of aflatoxin B1 to Bifidobacteria in vitro. J. Food Prot. 2000, 63, 1133–1136. [Google Scholar] [CrossRef]
- Karazhyan, R.; Shaker Sheyda, I.; Mehraban Sang Atash, M.; Tajalli, F.; Mojtahedi, M.; Sadegh, M. Effect of Saccharomyces cerevisiae yeast on ruminal detoxification of aflatoxin B1. J. Vet. Res. 2017, 72, 81–86. [Google Scholar]
- El-Nezami, H.; Kankaanpää, P.; Salminen, S.; Ahokas, J. Physicochemical alterations enhance the ability of dairy strains of lactic acid bacteria to remove aflatoxin from contaminated media. J. Food Prot. 1998, 61, 466–468. [Google Scholar] [CrossRef] [PubMed]
- El-Nezami, H.; Polychronaki, N.; Salminen, S.; Mykkänen, H. Binding rather than metabolism may explain the interaction of two food-grade Lactobacillus strains with zearalenone and its derivative α-zearalenol. Appl. Environ. Microbiol. 2002, 68, 3545–3549. [Google Scholar] [CrossRef] [PubMed]
- El-Nezami, H.; Polychronaki, N.; Lee, Y.K.; Haskard, C.; Juvonen, R.; Salminen, S.; Mykkänen, H. Chemical moieties and interactions involved in the binding of zearalenone to the surface of Lactobacillus rhamnosus strains GG. J. Agric. Food Chem. 2004, 52, 4577–4581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.G.; Liu, S.; Zhao, X.J.; Wang, N.; Jiang, X.; Xin, H.S.; Zhang, Y.G. Lactobacillus rhamnosus GG modulates gastrointestinal absorption, excretion patterns, and toxicity in Holstein calves fed a single dose of aflatoxin B1. J. Dairy Sci. 2018, 102, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, K.H.; Pettersson, H.; Sandholm, K.; Olsen, M. Metabolism of aflatoxin, ochratoxin, zearalenone, and three trichothecenes by intact rumen fluid, rumen protozoa, and rumen bacteria. Appl. Environ. Microbiol. 1984, 47, 1070–1073. [Google Scholar] [PubMed]
- Gallo, A.; Giuberti, G.; Bertuzzi, T.; Moschini, M.; Masoero, F. Study of the effects of PR toxin, mycophenolic acid and roquefortine C on in vitro gas production parameters and their stability in the rumen environment. J. Agric. Sci. 2015, 153, 163–176. [Google Scholar] [CrossRef]
- Fink-Gremmels, J. Mycotoxins in cattle feeds and carry-over to dairy milk: A review. Food Addit. Contam. Part A 2008, 25, 172–180. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.S.; Lee, J.H.; Simizu, Y.; Tazaki, H.; Itabashi, H.; Kimura, N. Effects of the Fusarium mycotoxin deoxynivalenol on in vitro rumen fermentation. Anim. Feed Sci. Technol. 2010, 162, 144–148. [Google Scholar] [CrossRef]
- Dänicke, S.; Matthäus, K.; Lebzien, P.; Valenta, H.; Stemme, K.; Ueberschär, K.-H.; Razzazi-Fazeli, E.; Böhm, J.; Flachowsky, G. Effects of Fusarium toxin-contaminated wheat grain on nutrient turnover, microbial protein synthesis and metabolism of deoxynivalenol and zearalenone in the rumen of dairy cows. J. Anim. Physiol. Anim. Nutr. (Berl.) 2005, 89, 303–315. [Google Scholar] [CrossRef]
- Seeling, K.; Dänicke, S.; Ueberschär, H.; Lebzien, P.; Flachowsky, G. On the effects of Fusarium toxin-contaminated wheat and the feed intake level on the metabolism and carry over of zearalenone in dairy cows. Food Addit. Contam. 2005, 22, 847–855. [Google Scholar] [CrossRef]
- Kennedy, D.G.; Hewitt, S.A.; McEvoy, J.D.G.; Currie, J.W.; Cannavan, A.; Blanchflower, W.J.; Elliot, C.T. Zeranol is formed from Fusarium spp. toxins in cattle in vivo. Food Addit. Contam. 1998, 15, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Songsermsakul, E.; Razzazi-Fazeli, P. A review of recent trends in applications of liquid chromatography-mass spectrometry for determination of mycotoxins. J. Liq. Chromatogr. Relat. Technol. 2008, 31, 1641–1686. [Google Scholar] [CrossRef]
- De Baere, S.; Osselaere, A.; Devreese, M.; Vanhaecke, L.; De Backer, P.; Croubels, S. Development of a liquid-chromatography tandem mass spectrometry and ultra-high-performance liquid chromatography high-resolution mass spectrometry method for the quantitative determination of zearalenone and its major metabolites in chicken and pig plasma. Anal. Chim. Acta 2012, 756, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Breidbach, A. A greener, quick and comprehensive extraction approach for LC-MS of multiple mycotoxins. Toxins (Basel) 2017, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Gałuszka, A.; Migaszewski, Z.; Namieśnik, J. The 12 principles of green analytical chemistry and the SIGNIFICANCE mnemonic of green analytical practices. Trends Anal. Chem. 2013, 50, 78–84. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Veterinary Use (CVMP). VICH Topic GL49: Studies to Evaluate the Metabolism and Residue Kinetics of Veterinary Drugs in Food Producing Animals: Validation of Analytical Methods Used in Residue Depletion Studies; Committee for Medicinal Products for Veterinary Use: London, UK, 2015. [Google Scholar]
- Ye, J.; Wu, Y.; Wang, S.; Guo, Q.; Lu, M.; Wu, C.; Chen, Y. Simultaneous determination of 16 mycotoxins in cereals using an Agilent Triple Quadrupole LC/MS system and e-Method. Agil. Technol. 2017, 2017, 1–10. [Google Scholar]
- Frenich, A.G.; Romero-González, R.; Gómez-Pérez, M.L.; Vidal, J.L.M. Multi-mycotoxin analysis in eggs using a QuEChERS-based extraction procedure and ultra-high-pressure liquid chromatography coupled to triple quadrupole mass spectrometry. J. Chromatogr. A 2011, 1218, 4349–4356. [Google Scholar] [CrossRef]
- Rasmussen, R.R.; Storm, I.M.L.D.; Rasmussen, P.H.; Smedsgaard, J.; Nielsen, K.F. Multi-mycotoxin analysis of maize silage by LC-MS/MS. Anal. Bioanal. Chem. 2010, 397, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Molinié, A.; Faucet, V.; Castegnaro, M.; Pfohl-Leszkowicz, A. Analysis of some breakfast cereals on the French market for their contents of ochratoxin A, citrinin and fumonisin B1: Development of a method for simultaneous extraction of ochratoxin A and citrinin. Food Chem. 2005, 92, 391–400. [Google Scholar] [CrossRef]
- Hu, L.; Rychlik, M. Biosynthesis of 15N3-labeled enniatins and beauvericin and their application to stable isotope dilution assays. J. Agric. Food Chem. 2012, 60, 7129–7136. [Google Scholar] [CrossRef]
- European Commission. Commission Recommendation No 2006/576/EC of 17 August 2006 on the presence of deoxynivalenol, zearalenone, ochratoxin A, T-2 and HT-2 and fumonisins in products intended for animal feeding. Off. J. Eur. Union 2006, 229, 7–9. [Google Scholar]
- European Commission. Commission Decision 2002/657/EC implementing Council Directive 96/23/EC concerning the performances of analytical methods and the interpretation of results. Off. J. Eur. Communities 2002, 221, 8–36. [Google Scholar]
- Knecht, J.; Stork, G. Prozentuales und logarithmisches verfahren zur berechnung von eichkurven. Z. Anal. Chem. 1974, 270, 97–98. [Google Scholar] [CrossRef]
- Heitzman, R. Veterinary Drug Residues Residues in Food Producing Animals and Their Products: Reference Materials and Methods; Blackwell Scientific Publications: Oxford, UK, 1992; Volume 636, ISBN 0-8412-3419-1. [Google Scholar]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Precursor Ion (m/z) a | Product Ions (m/z) | CV b (V) | CE c (eV) | RT d (min) |
|---|---|---|---|---|---|
| DON | 297.2 [M+H]+ | 248.9 231.0 | 20 | 10 12 | 4.06 |
| 13C15-DON | 312.0 [M+H]+ | 263.0 245.2 | 20 | 10 10 | 4.06 |
| DOM-1 | 281.0 [M+H]+ | 233.0 215.0 | 27 | 10 10 | 4.66 |
| NIV | 313.1 [M+H]+ | 175.0 | 35 | 15 | 3.49 |
| ENN B | 640.2 [M+H]+ | 196.2 213.8 | 70 | 20 22 | 8.04 |
| 15N3-ENN B | 643.3 [M+H]+ | 197.1 215.3 | 70 | 23 23 | 8.04 |
| MPA | 321.1 [M+H]+ | 207.0 159.0 | 25 | 22 33 | 6.43 |
| 13C17-MPA | 338.0 [M+H]+ | 320.0 218.0 | 26 | 10 22 | 6.43 |
| ROQ-C | 390.2 [M+H]+ | 192.9 322.0 | 32 | 25 20 | 5.94 |
| 13C22-ROQ-C | 412.0 [M+H]+ | 201.0 339.0 | 32 | 27 18 | 5.94 |
| ZEN | 317.3 [M−H]− | 175.0 131.0 | 15 | 25 30 | 4.95 |
| 13C18-ZEN | 335.3 [M−H]− | 185.1 169.1 | 15 | 25 32 | 4.95 |
| ZAN | 319.2 [M−H]− | 275.2 205.1 | 20 | 20 22 | 4.90 |
| α-ZEL | 319.2 [M−H]− | 275.2 301.0 | 20 | 20 22 | 4.37 |
| β-ZEL | 319.2 [M−H]− | 275.2 301.0 | 20 | 20 22 | 3.99 |
| α-ZAL | 321.2 [M−H]− | 277.1 303.1 | 30 | 22 20 | 4.29 |
| β-ZAL | 321.8 [M−H]− | 277.1 303.1 | 30 | 22 20 | 3.92 |
| Analyte(s) ESI+ | Time (min) | MP A:B Ratio (v:v) |
| DON, 13C15-DON, DOM-1, NIV, ENN B, 15N3-ENN B, MPA, 13C17-MPA, ROQ-C, 13C22-ROQ-C | 0.00–0.50 0.50–5.50 5.50–7.50 7.50–7.70 7.70–10.0 | 95:5 Linear to 5:95 5:95 Linear to 95:5 95:5 |
| Analyte(s) ESI− | Time (min) | MP C:D Ratio (v:v) |
| ZEN, 13C18-ZEN, ZAN, α-ZEL, β-ZEL, α-ZAL, β-ZAL | 0.00–0.50 0.50–3.50 3.50–4.90 4.90–5.00 5.00–7.00 | 70:30 Linear to 30:70 30:70 Linear to 70:30 70:30 |
| Analyte | Calibration Curve Range (ng/mL) | LOD (ng/mL) | LOQ (ng/mL) | Medium (ng/mL) | High (ng/mL) | r | GOF (%) |
|---|---|---|---|---|---|---|---|
| DON | 0.45–180 | 0.05 | 0.45 | 12 | 120 | 0.9996 | 4.83 |
| DOM-1 | 1.56–180 | 0.08 | 1.56 | 12 | 120 | 0.9995 | 4.67 |
| NIV | 36–600 | 5.43 | 36 | 120 | 600 | 0.9991 | 4.44 |
| ENN B | 0.39–15 | <0.01 | 0.39 | 1.4 | 10 | 0.9982 | 6.36 |
| MPA | 0.6–90 | 0.17 | 0.60 | 6.0 | 60 | 0.9995 | 8.93 |
| ROQ-C | 0.1–30 | <0.01 | 0.10 | 2.0 | 20 | 0.9996 | 5.29 |
| ZEN | 0.3–45 | 0.02 | 0.30 | 3.0 | 30 | 0.9993 | 7.82 |
| ZAN | 0.3–45 | 0.07 | 0.30 | 3.0 | 30 | 0.9997 | 5.02 |
| α-ZEL | 0.3–45 | 0.08 | 0.30 | 3.0 | 30 | 0.9990 | 6.24 |
| β-ZEL | 0.3–45 | 0.07 | 0.30 | 3.0 | 30 | 0.9938 | 7.78 |
| α-ZAL | 0.3–45 | 0.02 | 0.30 | 3.0 | 30 | 0.9983 | 4.82 |
| β-ZAL | 0.3–45 | 0.02 | 0.30 | 3.0 | 30 | 0.9931 | 11.15 |
| Analyte | Within-Run (n = 6) | Between-Run (n = 2 × 3) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Accuracy (%) | Precision (RSD, %) | Accuracy (%) | Precision (RSD, %) | |||||||||
| LOQ | Medium | High | LOQ | Medium | High | LOQ | Medium | High | LOQ | Medium | High | |
| DON | 19.4 A | −0.8 C | 1.0 C | 5.2 A | 2.8 C | 1.6 D | 19.5 A | −1.6 C | 0.4 C | 5.2 | 2.5 | 1.8 |
| DOM-1 | 3.7 B | −1.8 C | −5.9 C | 5.5 B | 5.2 C | 9.2 D | 2.6 B | −4.7 C | −2.9 C | 5.2 | 5.8 | 6.6 |
| NIV | 4.2 C | −7.6 C | −7.2 C | 5.0 C | 7.6 D | 7.7 D | 1.8 C | −8.7 C | −6.1 C | 6.8 | 7.8 | 6.5 |
| ENN B | −14.0 A | 3.6 B | −5.8 C | 5.0 A | 2.0 B | 2.2 C | −14.0 A | 1.1 B | −3.8 C | 5.0 | 2.4 | 3.9 |
| MPA | 3.2 A | −8.2 B | 5.2 C | 5.3 A | 2.9 B | 4.5 C | 4.5 A | −5.1 B | 0.5 C | 15.5 | 4.7 | 5.8 |
| ROQ-C | 4.7 A | −2.5 B | −4.9 C | 4.5 A | 2.0 B | 1.3 C | 6.8 A | −3.1 B | −4.5 C | 4.2 | 2.4 | 2.5 |
| ZEN | −5.6 A | 2.0 B | −3.9 C | 16.6 A | 6.2 B | 1.5 C | −0.3 A | −3.1 B | 0.5 C | 10.3 | 4.5 | 2.4 |
| ZAN | 3.6 A | −2.4 B | −6.5 C | 11.1 A | 5.2 B | 3.9 C | -0.6 A | −0.5 B | −2.0 C | 12.4 | 4.4 | 5.3 |
| α-ZEL | −7.6 A | −2.4 B | −10.6 C | 9.4 A | 5.9 B | 1.1 C | −9.2 A | −0.6 B | −5.1 C | 12.4 | 5.9 | 8.2 |
| β-ZEL | −4.6 A | −19.3 B | 4.6 C | 8.6 A | 12.0 B | 6.3 C | −2.8 A | −8.6 B | −5.8 C | 8.2 | 13.4 | 14.5 |
| α-ZAL | −21.3 A | −5.0 B | 1.8 C | 14.5 A | 9.2 B | 2.9 C | −13.5 A | −1.9 B | −5.5 C | 16.9 | 10.1 | 9.6 |
| β-ZAL | −6.7 A | −10.2 B | −9.9 C | 16.4 A | 12.0 B | 10.9 C | −6.9 A | −5.3 B | −7.1 C | 16.8 | 14.3 | 14.9 |
| Analyte | Matrix A: RF | Matrix B: RF + MS | ||
|---|---|---|---|---|
| SSE (%) | RE (%) | SSE (%) | RE (%) | |
| DON | 79 | 42 | 55 | 44 |
| NIV | 68 | 9 | 60 | 8 |
| ENN B | 241 | 13 | 197 | 16 |
| MPA | 45 | 2 | 48 | 3 |
| ROQ-C | 69 | 16 | 62 | 16 |
| ZEN | 60 | 19 | 52 | 19 |
| DOM-1 | 71 | 46 | 60 | 48 |
| α-ZEL | 60 | 20 | 53 | 21 |
| β-ZEL | 67 | 21 | 60 | 25 |
| ZAN | 64 | 19 | 53 | 21 |
| α-ZAL | 64 | 22 | 58 | 25 |
| β-ZAL | 68 | 23 | 63 | 29 |
| Analyte | Calibration Curve Range (ng/mL) | LOD (ng/mL) | LOQ (ng/mL) | Medium (ng/mL) | High (ng/mL) | r | GOF (%) |
|---|---|---|---|---|---|---|---|
| DON | 8–187 | 0.41 | 7.99 | 19.4 | 120 | 0.9998 | 1.65 |
| DOM-1 | 6–180 | 0.35 | 6.00 | 12.0 | 120 | 0.9993 | 3.22 |
| NIV | 36–900 | 9.28 | 36 | 120 | 600 | 0.9974 | 6.64 |
| ENN B | 0.44–15.34 | 0.01 | 0.44 | 1.34 | 10 | 0.9985 | 5.81 |
| MPA | 1.20–90 | 0.42 | 1.20 | 6.0 | 60 | 0.9986 | 6.55 |
| ROQ-C | 0.2–30 | 0.01 | 0.20 | 2.0 | 20 | 0.9978 | 9.29 |
| ZEN | 1.06–45 | 0.04 | 1.06 | 3.5 | 30 | 0.9995 | 4.09 |
| ZAN | 0.3–45 | 0.10 | 0.30 | 3.0 | 30 | 0.9989 | 5.58 |
| α-ZEL | 0.6–45 | 0.15 | 0.60 | 3.0 | 30 | 0.9994 | 5.24 |
| β-ZEL | 0.6–45 | 0.08 | 0.60 | 3.0 | 30 | 0.9992 | 6.57 |
| α-ZAL | 1.5–45 | 0.04 | 1.50 | 3.0 | 30 | 0.9993 | 4.91 |
| β-ZAL | 0.3–45 | 0.06 | 0.30 | 3.0 | 30 | 0.9994 | 5.83 |
| Analyte | Within-Run (n = 6) | |||||
|---|---|---|---|---|---|---|
| Accuracy (%) | Precision (RSD, %) | |||||
| LOQ | Medium | High | LOQ | Medium | High | |
| DON | −0.3 B | 0.1 C | −4.8 C | 0.6 B | 2.5 C | 3.6 D |
| DOM-1 | −7.9 B | −6.3 C | 1.9 C | 9.7 B | 3.3 C | 4.7 D |
| NIV | 1.1 C | 5.5 C | −1.0 C | 5.6 C | 2.8 D | 8.0 D |
| ENN B | −2.0 A | 4.5 B | −8.5 C | 4.0 A | 2.9 B | 5.1 C |
| MPA | −1.1 B | −7.3 B | 3.6 C | 5.0 B | 8.9 B | 7.4 C |
| ROQ-C | −12.5 A | −1.0 B | −5.2 C | 3.1 A | 3.3 B | 3.0 C |
| ZEN | −6.0 B | 2.5 B | 3.1 C | 8.8 B | 6.2 B | 7.3 C |
| ZAN | −2.2 A | 0.9 B | 8.3 C | 6.0 A | 4.7 B | 0.9 C |
| α-ZEL | 1.9 A | −7.6 B | −5.2 C | 4.7 A | 2.9 B | 6.5 C |
| β-ZEL | −1.4 A | −17.6 B | −0.7 C | 8.2 A | 10.0 B | 7.8 C |
| α-ZAL | −10.9 B | −17.0 B | 7.4 C | 7.8 B | 9.5 B | 6.7 C |
| β-ZAL | −8.3 A | −16.8 B | −2.9 C | 6.4 A | 11.1 B | 7.5 C |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Debevere, S.; De Baere, S.; Haesaert, G.; Rychlik, M.; Fievez, V.; Croubels, S. Development of an UPLC-MS/MS Method for the Analysis of Mycotoxins in Rumen Fluid with and without Maize Silage Emphasizes the Importance of Using Matrix-Matched Calibration. Toxins 2019, 11, 519. https://doi.org/10.3390/toxins11090519
Debevere S, De Baere S, Haesaert G, Rychlik M, Fievez V, Croubels S. Development of an UPLC-MS/MS Method for the Analysis of Mycotoxins in Rumen Fluid with and without Maize Silage Emphasizes the Importance of Using Matrix-Matched Calibration. Toxins. 2019; 11(9):519. https://doi.org/10.3390/toxins11090519
Chicago/Turabian StyleDebevere, Sandra, Siegrid De Baere, Geert Haesaert, Michael Rychlik, Veerle Fievez, and Siska Croubels. 2019. "Development of an UPLC-MS/MS Method for the Analysis of Mycotoxins in Rumen Fluid with and without Maize Silage Emphasizes the Importance of Using Matrix-Matched Calibration" Toxins 11, no. 9: 519. https://doi.org/10.3390/toxins11090519
APA StyleDebevere, S., De Baere, S., Haesaert, G., Rychlik, M., Fievez, V., & Croubels, S. (2019). Development of an UPLC-MS/MS Method for the Analysis of Mycotoxins in Rumen Fluid with and without Maize Silage Emphasizes the Importance of Using Matrix-Matched Calibration. Toxins, 11(9), 519. https://doi.org/10.3390/toxins11090519