Characterization of Phase I and Glucuronide Phase II Metabolites of 17 Mycotoxins Using Liquid Chromatography—High-Resolution Mass Spectrometry


Abstract
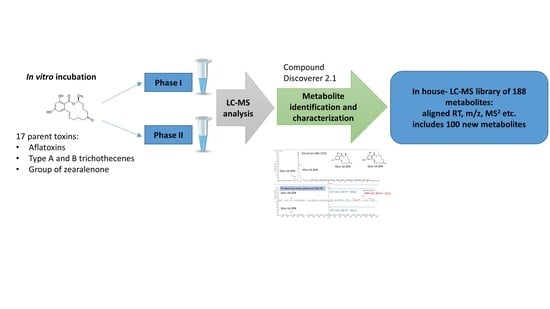
:
1. Introduction
2. Results
2.1. Trichothecene Type A and B
2.1.1. Trichothecene Type A
2.1.2. Trichothecene Type B
2.2. Aflatoxins
2.3. Group of Zearalenone
4. Conclusions
5. Materials and Methods
5.1. Chemicals
5.2. Mycotoxin Standards
5.3. Experimental Design and Microsomal Incubations
5.4. LC-HRMS Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alshannaq, A.; Yu, J. Occurrence, Toxicity, and Analysis of Major Mycotoxins in Food. Int. J. Environ. Res. Public Health 2017, 14, 632. [Google Scholar] [CrossRef] [PubMed]
- Roscoe, V.; Lombaert, G.; Huzel, V.; Neumann, G.; Melietio, J.; Kitchen, D.; Kotello, S.; Krakalovich, T.; Trelka, R.; Scott, P. Mycotoxins in breakfast cereals from the Canadian retail market: A 3-year survey. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2008, 25, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, A.-M.; Scott, P. Ochratoxin A in cocoa and chocolate sampled in Canada. Food Addit. Contam. Part A 2011, 28, 762–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolakowski, B.; O’Rourke, S.M.; Bietlot, H.P.; Kurz, K.; Aweryn, B. Ochratoxin A Concentrations in a Variety of Grain-Based and Non – Grain-Based Foods on the Canadian Retail Market from 2009 to 2014. J. Food Prot. 2016, 79, 2143–2159. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.; Mankotia, M.; Pantazopoulos, P.; Neil, R.J.; Scott, P.M. Ochratoxin A in wine and grape juice sold in Canada. Food Addit. Contam. 2004, 21, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Lombaert, G.A.; Pellaers, P.; Roscoe, V.; Mankotia, M.; Neil, R.; Scott, P.M. Mycotoxins in infant cereal foods from the Canadian retail market. Food Addit.Contam 2003, 20, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Hooker, D.C.; Schaafsma, A.W. Agronomic and environmental impacts on concentrations of deoxynivalenol and fumonisin B1 in corn across Ontario. Can. J. Plant Pathol. 2005, 27, 347–356. [Google Scholar] [CrossRef]
- Schatzmayr, G.; Streit, E. Global occurrence of mycotoxins in the food and feed chain: Facts and figures. World Mycotoxin J. 2013, 6, 213–222. [Google Scholar] [CrossRef]
- Canadian Food Inspection Agency. 2013–2015 Multi-Mycotoxin Analysis in Selected Foods; Canadian Food Inspection Agency: Ottawa, ON, Canada, 2016. [Google Scholar]
- Smith, M.; Madec, S.; Coton, E.; Hymery, N. Natural Co-Occurrence of Mycotoxins in Foods and Feeds and Their in vitro Combined Toxicological Effects. Toxins 2016, 8, 94. [Google Scholar] [CrossRef]
- Serrano, A.B.; Font, G.; Ruiz, M.J.; Ferrer, E. Co-occurrence and risk assessment of mycotoxins in food and diet from Mediterranean area. Food Chem. 2012, 135, 423–429. [Google Scholar] [CrossRef]
- Wells, L.; Hardie, L.; Williams, C.; White, K.; Liu, Y.; De Santis, B.; Debegnach, F.; Moretti, G.; Greetham, S.; Brera, C.; et al. Deoxynivalenol Biomarkers in the Urine of UK Vegetarians. Toxins 2017, 9, 196. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-carrasco, Y.; Moltó, J.C.; Mañes, J.; Berrada, H. Exposure assessment approach through mycotoxin/creatinine ratio evaluation in urine by GC—MS/MS. Food Chem. Toxicol. 2014, 72, 69–75. [Google Scholar]
- Solfrizzo, M.; Gambacorta, L.; Lattanzio, V.M.T.; Powers, S.; Visconti, A. Simultaneous LC-MS/MS determination of aflatoxin M 1, ochratoxin A, deoxynivalenol, de-epoxydeoxynivalenol, α and β-zearalenols and fumonisin B 1 in urine as a multi-biomarker method to assess exposure to mycotoxins. Anal. Bioanal. Chem. 2011, 401, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Heyndrickx, E.; Sioen, I.; Huybrechts, B.; Callebaut, A.; De Henauw, S.; De Saeger, S. Human biomonitoring of multiple mycotoxins in the Belgian population: Results of the BIOMYCO study. Environ. Int. 2015, 84, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.C.; Burley, V.J.; Rothwell, J.A.; White, K.L.M.; Cade, J.E.; Wild, C.P. Deoxynivalenol: Rationale for development and application of a urinary biomarker. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2008, 25, 864–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, A.; Claeys, L.; Mengelers, M.; Vanhoorne, V.; Vervaet, C.; Huybrechts, B.; De Saeger, S.; De Boevre, M. Humans significantly metabolize and excrete the mycotoxin deoxynivalenol and its modified form deoxynivalenol-3-glucoside within 24 hours. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Maul, R.; Warth, B.; Kant, J.S.; Schebb, N.H.; Krska, R.; Koch, M.; Sulyok, M. Investigation of the hepatic glucuronidation pattern of the Fusarium mycotoxin deoxynivalenol in various species. Chem. Res. Toxicol. 2012, 25, 2715–2717. [Google Scholar] [CrossRef] [PubMed]
- Warth, B.; Sulyok, M.; Fruhmann, P.; Berthiller, F.; Schuhmacher, R.; Hametner, C.; Adam, G.; Fröhlich, J.; Krska, R. Assessment of human deoxynivalenol exposure using an LC-MS/MS based biomarker method. Toxicol. Lett. 2012, 211, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.; Mengelers, M.; Yang, S.; De Saeger, S.; De Boevre, M. Mycotoxin Biomarkers of Exposure: A Comprehensive Review. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1127–1155. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Carrasco, Y.; Moltó, J.C.; Mañes, J.; Berrada, H. Development of microextraction techniques in combination with GC–MS/MS for the determination of mycotoxins and metabolites in human urine. J. Sep. Sci. 2017, 40, 1572–1582. [Google Scholar] [CrossRef]
- Föllmann, W.; Ali, N.; Blaszkewicz, M.; Degen, G.H. Biomonitoring of Mycotoxins in Urine: Pilot Study in Mill Workers. J. Toxicol. Environ. Health. A 2016, 79, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Li, X.; Li, J.; Niu, Y.; Shi, L.; Fang, Z.; Zhang, T.; Ding, H. Quantitative determination of carcinogenic mycotoxins in human and animal biological matrices and animal-derived foods using multi-mycotoxin and analyte-specific high performance liquid chromatography-tandem mass spectrometric methods. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1073, 191–200. [Google Scholar] [CrossRef] [PubMed]
- De Santis, B.; Raggi, M.E.; Moretti, G.; Facchiano, F.; Mezzelani, A.; Villa, L.; Bonfanti, A.; Campioni, A.; Rossi, S.; Camposeo, S.; et al. Study on the association among mycotoxins and other variables in children with autism. Toxins 2017, 9, 203. [Google Scholar] [CrossRef] [PubMed]
- Escrivá, L.; Manyes, L.; Font, G.; Berrada, H. Mycotoxin analysis of human urine by LC-MS/MS: A comparative extraction study. Toxins 2017, 9, 330. [Google Scholar] [CrossRef] [PubMed]
- Warth, B.; Sulyok, M.; Fruhmann, P.; Mikula, H.; Berthiller, F.; Schuhmacher, R.; Hametner, C.; Abia, W.A.; Adam, G.; Fröhlich, J.; et al. Development and validation of a rapid multi-biomarker liquid chromatography/tandem mass spectrometry method to assess human exposure to mycotoxins. Rapid Commun. Mass Spectrom. 2012, 26, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Ediage, E.N.; Di Mavungu, J.D.; Song, S.; Wu, A.; Van Peteghem, C.; De Saeger, S. A direct assessment of mycotoxin biomarkers in human urine samples by liquid chromatography tandem mass spectrometry. Anal. Chim. Acta 2012, 741, 58–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huybrechts, B.; Martins, J.C.; Debongnie, P.; Uhlig, S.; Callebaut, A. Fast and sensitive LC–MS/MS method measuring human mycotoxin exposure using biomarkers in urine. Arch. Toxicol. 2015, 89, 1993–2005. [Google Scholar] [CrossRef] [PubMed]
- Slobodchikova, I.; Vuckovic, D. Liquid chromatography – high resolution mass spectrometry method for monitoring of 17 mycotoxins in human plasma for exposure studies. J. Chromatogr. A 2018, 1548, 51–63. [Google Scholar] [CrossRef]
- Osteresch, B.; Viegas, S.; Cramer, B.; Humpf, H.U. Multi-mycotoxin analysis using dried blood spots and dried serum spots. Anal. Bioanal. Chem. 2017, 409, 3369–3382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Zhang, H.; Sun, F.; De Ruyck, K.; Zhang, J.; Jin, Y.; Li, Y.; Wang, Z.; Zhang, S.; De Saeger, S.; et al. Metabolic Profile of Zearalenone in Liver Microsomes from Different Species and Its in Vivo Metabolism in Rats and Chickens Using Ultra High-Pressure Liquid Chromatography-Quadrupole/Time-of-Flight Mass Spectrometry. J. Agric. Food Chem. 2017, 65, 11292–11303. [Google Scholar] [CrossRef]
- Yang, S.; DE Boevre, M.; Li, Y.; De Saeger, S. The Toxicokinetics of HT-2 Toxin in Rats and Its Metabolic Profile in Livestock and Human Liver Microsomes. J. Agric. Food Chem. 2018, 66, 8160–8168. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; De Boevre, M.; Zhang, H.; De Ruyck, K.; Sun, F.; Zhang, J.; Jin, Y.; Li, Y.; Wang, Z.; Zhang, S.; et al. Metabolism of T-2 Toxin in Farm Animals and Human In Vitro and in Chickens In Vivo Using Ultra High-Performance Liquid Chromatography- Quadrupole/Time-of-Flight Hybrid Mass Spectrometry Along with Online Hydrogen/Deuterium Exchange Technique. J. Agric. Food Chem. 2017, 65, 7217–7227. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, E.; Hildebrand, A.; Mikula, H.; Metzler, M. Glucuronidation of zearalenone, zeranol and four metabolites in vitro: Formation of glucuronides by various microsomes and human UDP-glucuronosyltransferase isoforms. Mol. Nutr. Food Res. 2010, 54, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.E.; Hansen, R.P.; Loader, J.I.; Jensen, D.J.; Cooney, J.M.; Wilkins, A.L.; Miles, C.O. Preparative Enzymatic Synthesis of Glucuronides of Zearalenone and Five of Its Metabolites. J. Agric. Food Chem. 2008, 56, 4032–4038. [Google Scholar] [CrossRef] [PubMed]
- Frizzell, C.; Uhlig, S.; Miles, C.O.; Verhaegen, S.; Elliott, C.T.; Eriksen, G.S.; Sørlie, M.; Ropstad, E.; Connolly, L. Biotransformation of zearalenone and zearalenols to their major glucuronide metabolites reduces estrogenic activity. Toxicol. Vitr. 2015, 29, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Mu, P.; Deng, Y. Mycotoxins: Cytotoxicity and biotransformation in animal cells. Toxicol. Res. 2016, 5, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Yanni, S.; Annaert, P.P.; Augustijns, P.; Bridges, A.; Gao, Y.; Daniel, K.; Thakker, D.R. Role of Flavin-Containing Monooxygenase in Oxidative Metabolism of Voriconazole by Human Liver Microsomes. Drug Metab. Dispos. 2009, 36, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Grothusen, A.; Hardt, J.; Bräutigam, L.; Lang, D.; Böcker, R. A convenient method to discriminate between cytochrome P450 enzymes and flavin containing monooxygenases in human liver microsomes. Arch. Toxicol. 1996, 71, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, R.; Nakajima, M.; Yamamoto, T.; Nagao, H.; Yokoi, T. In silico and in vitro Approaches to Elucidate the Thermal Stability of Human UDP-glucuronosyltransferase UGT 1A9. Drug Metab. Pharmacokinet. 2009, 24, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, Y.; Cao, X.; Hu, D.; Wang, Z.; Wang, Y.; Shen, J.; Zhang, S. Metabolic pathways of T-2 toxin in in vivo and in vitro systems of Wistar rats. J. Agric. Food Chem. 2013, 61, 9734–9743. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, G.; Zhao, H.; Zheng, J.; Hu, F.; Fang, B. Liquid chromatography-tandem mass spectrometry method for toxicokinetics, tissue distribution, and excretion studies of T-2 toxin and its major metabolites in pigs. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 958, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Welsch, T.; Humpf, H.U. HT-2 toxin 4-glucuronide as new T-2 toxin metabolite: Enzymatic synthesis, analysis, and species specific formation of T-2 and HT-2 toxin glucuronides by rat, mouse, pig, and human liver microsomes. J. Agric. Food Chem. 2012, 60, 10170–10178. [Google Scholar] [CrossRef] [PubMed]
- Ajandouz, E.H.; Berdah, S.; Moutardier, V.; Bege, T.; Birnbaum, D.J.; Perrier, J.; Di Pasquale, E.; Maresca, M. Hydrolytic Fate of 3/15-Acetyldeoxynivalenol in Humans: Specific Deacetylation by the Small Intestine and Liver Revealed Using in Vitro and ex Vivo Approaches. Toxins 2016, 8, 232. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, G.S.; Pettersson, H. Lack of de-epoxidation of type B trichothecenes in incubates with human faeces. Food Addit. Contam. 2003, 20, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Schwartz-Zimmermann, H.E.; Binder, S.B.; Hametner, C.; Miró-Abella, E.; Schwarz, C.; Michlmayr, H.; Reiterer, N.; Labudova, S.; Adam, G.; Berthiller, F. Metabolism of nivalenol and nivalenol-3-glucoside in rats. Toxicol. Lett. 2019, 306, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Gratz, S.W.; Duncan, G.; Richardson, A.J. The Human Fecal Microbiota Metabolizes Deoxynivalenol and Deoxynivalenol-3-Glucoside and May Be Responsible for Urinary. Appl. Environ. Microbiol. 2013, 79, 1821–1825. [Google Scholar] [CrossRef]
- Brezina, U.; Rempe, I.; Kersten, S.; Valenta, H.; Humpf, H.U.; Dänicke, S. Diagnosis of intoxications of piglets fed with Fusarium toxin-contaminated maize by the analysis of mycotoxin residues in serum, liquor and urine with LC-MS/MS. Arch. Anim. Nutr. 2014, 68, 425–447. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P.; Arneson, K.O.; Williams, K.M.; Deng, Z.; Harris, T.M. Reaction of aflatoxin B1 oxidation products with lysine. Chem. Res. Toxicol. 2002, 15, 780–792. [Google Scholar] [CrossRef]
- Johnson, W.W.; Guengerich, F.P. Reaction of aflatoxin B1 exo-8,9-epoxide with DNA: Kinetic analysis of covalent binding and DNA-induced hydrolysis. Proc. Natl. Acad. Sci. USA 1997, 94, 6121–6125. [Google Scholar] [CrossRef]
- Everley, R.A.; Ciner, F.L.; Zhang, D.; Scholl, P.F.; Groopman, J.D.; Croley, T.R. Measurement of aflatoxin and aflatoxin metabolites in urine by liquid chromatography-tandem mass spectrometry. J. Anal. Toxicol. 2007, 31, 150–156. [Google Scholar] [CrossRef]
- Walton, M.; Egner, P.; Scholl, P.F.; Walker, J.; Kensler, T.W.; Groopman, J.D. Liquid chromatography electrospray-mass spectrometry of urinary aflatoxin biomarkers: Characterization and application to dosimetry and chemoprevention in rats. Chem. Res. Toxicol. 2001, 14, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Andrade, P.D.; Gomas da Silva, J.L.; Caldas, E.D. Simultaneous analysis of aflatoxins B1, B2, G1, G2, M1 and ochratoxin A in breast milk by high-performance liquid chromatography/fluorescence after liquid-liquid extraction with low temperature purification (LLE-LTP). J. Chromatogr. A 2013, 1304, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Li, Q.; Sun, L.; Du, Y.; Xia, J.; Zhang, Y. Simultaneous determination of aflatoxin B 1 and M 1 in milk, fresh milk and milk powder by LC-MS/MS utilising online turbulent flow chromatography. Food Addit. Contam. Part A 2015, 32, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Leppänen, J.M.; Partanen, H.A.; Vähäkangas, K.H.; Woodhouse, H.J.; Myllynen, P.K.; El-Nezami, H.S. Aflatoxin B1 Transfer and Metabolism in Human Placenta. Toxicol. Sci. 2009, 113, 216–225. [Google Scholar]
- Dohnal, V.; Wu, Q.; Kuc, K. Metabolism of aflatoxins: Key enzymes and interindividual as well as interspecies differences. Arch. Toxicol. 2014, 88, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- Jager, A.V.; Tonin, F.G.; Souto, P.C.M.C.; Privatti, R.T.; Oliveira, C.A.F. Determination of urinary biomarkers for assessment of short-term human exposure to aflatoxins in São Paulo, Brazil. Toxins 2014, 6, 1996–2007. [Google Scholar] [CrossRef] [PubMed]
- Solfrizzo, M.; Gambacorta, L.; Visconti, A. Assessment of multi-mycotoxin exposure in southern Italy by urinary multi-biomarker determination. Toxins 2014, 6, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Mykkänen, H.; Zhu, H.; Salminen, E.; Juvonen, R.O.; Ling, W.; Ma, J.; Polychronaki, N.; Kemiläinen, H.; Mykkänen, O.; Salminen, S.; et al. Fecal and urinary excretion of aflatoxin B1 metabolites (AFQ1, AFM1 and AFB-N7-guanine) in young Chinese males. Int. J. Cancer 2005, 115, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.P.; Wienkers, L.C.; Stapleton, P.L.; Kunze, K.L.; Eaton, D.L. Role of Human Microsomal and Human Complementary DNA-expressed Cytochromes P4501A2 and P4503A4 in the Bioactivation of Aflatoxin B11. Cancer Res. 1994, 54, 101–108. [Google Scholar]
- Wild, C.P.; Turner, P.C. The toxicology of aflatoxins as a basis for public health decisions. Mutagenesis 2002, 17, 471–481. [Google Scholar] [CrossRef]
- Diaz, G.J.; Cepeda, S.M.; Martos, P.A. Stability of aflatoxins in solution. J. AOAC Int. 2012, 95, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Bbosa, G.S.; Kitya, D.; Lubega, A.; Ogwal-Okeng, J.; Anokbonggo, W.W.; Kyegombe, D.B. Review of the Biological and Health Effects of Aflatoxins on Body Organs and Body Systems. Aflatoxins Recent Adv. Futur. Prospect. 2013, 12, 239–265. [Google Scholar]
- Hatem, N.L.; Hassab, H.M.A.; Abd Al-Rahman, E.M.; El-Deeb, S.A.; El-Sayed Ahmed, R.L. Prevalence of afl atoxins in blood and urine of Egyptian infants with protein – energy malnutrition. Food Nutr. Bull. 2005, 26, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Leong, Y.H.; Latiff, A.A.; Ahmad, N.I.; Rosma, A. Exposure measurement of aflatoxins and aflatoxin metabolites in human body fluids. A short review. Mycotoxin Res. 2012, 28, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Roebuck, B.D.; Siegel, W.G.; Wogan, G.N. In vitro metabolism of aflatoxin B2 by animal and human liver. Cancer Res. 1978, 38, 999–1002. [Google Scholar] [PubMed]
- Bravin, F.; Duca, R.C.; Balaguer, P.; Delaforge, M. In Vitro Cytochrome P450 Formation of a Mono-Hydroxylated Metabolite of Zearalenone Exhibiting Estrogenic Activities: Possible Occurrence of This Metabolite in Vivo. Int. J. Mol. Sci. 2009, 10, 1824–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrand, A.; Pfeiffer, E.; Metzler, M. Aromatic hydroxylation and catechol formation: A novel metabolic pathway of the growth promotor zeranol. Toxicol. Lett. 2010, 192, 379–386. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, H.; Zhang, J.; Li, Y.; Jin, Y.; Zhang, S.; De Saeger, S.; Li, Y.; Sun, F.; De Boevre, M. Deglucosylation of zearalenone-14-glucoside in animals and human liver leads to underestimation of exposure to zearalenone in humans. Arch. Toxicol. 2018, 92, 2779–2791. [Google Scholar] [CrossRef]
- Kluger, B.; Bueschl, C.; Neumann, N.; Stückler, R.; Doppler, M.; Chassy, A.W.; Waterhouse, A.L.; Rechthaler, J.; Kampleitner, N.; Thallinger, G.G.; et al. Untargeted profiling of tracer-derived metabolites using stable isotopic labeling and fast polarity-switching LC-ESI-HRMS. Anal. Chem. 2014, 86, 11533–11537. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mycotoxin | Oxidation Reactions and Number of Metabolites, (n) | ∑ n | ||||||
|---|---|---|---|---|---|---|---|---|
| Desaturation, oxidation, −(H4) +(O) | Desaturation, oxidation, −(H2) +(O) | Oxidation +(O) | Reduction, oxidation, +(H2) +(O) | Oxidation, +(O2) | Desaturation oxidation, −(H2) +(O2) | Oxidation, −(H4) +(O2) | ||
| ZEN | 0 | 1 | 9(75%) | 2 | 0 | 0 | 0 | 12 |
| α-ZOL | 1 | 6 | 8(53%) | 0 | 0 | 0 | 0 | 15 |
| β-ZOL | 1 | 1 | 7(88%) | 0 | 0 | 0 | 0 | 8 |
| ZAN | 0 | 1 | 5(23%) | 8(36%) | 6(27%) | 2 | 0 | 22 |
| α-ZAL | 1 | 3 | 4(40%) | 0 | 0 | 1 | 1 | 10 |
| β-ZAL | 1 | 4 | 8(53%) | 0 | 0 | 1 | 1 | 15 |
| Mycotoxin | Total Number of Glucuronides |
|---|---|
| ZEN | 10 |
| α-ZOL | 10 |
| β-ZOL | 7 |
| ZAN | 24 |
| α-ZAL | 10 |
| β-ZAL | 7 |
| Mycotoxins | Hydrolysis | Oxidation | De-Epoxidation | Epoxidation | Demethylation | Reduction | Glucuronidation |
|---|---|---|---|---|---|---|---|
| T-2 | ✓ | ✓ | ✓ | ||||
| HT-2 | ✓ | ✓ | ✓ | ||||
| 3-AcDON | ✓ | ✓ | |||||
| 15-AcDON | ✓ | ✓ | |||||
| FUS-X | ✓ | ✓ | |||||
| DON | ✓ | ✓ | |||||
| NIV | ✓ | ✓ | |||||
| AFB1 | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| AFB2 | ✓ | ||||||
| AFG1 | ✓ | ||||||
| AFG2 | ✓ | ||||||
| ZEN | ✓ | ✓ | |||||
| α-ZOL | ✓ | ✓ | |||||
| β-ZOL | ✓ | ✓ | |||||
| ZAN | ✓ | ✓ | |||||
| α-ZAL | ✓ | ✓ | |||||
| β-ZAL | ✓ | ✓ |
| Mycotoxin | Expected Metabolites | Missing Metabolites | LC-MS Library | New Metabolites |
|---|---|---|---|---|
| T-2 | Phase I metabolites: HT-2, 15-deacetyl-T-2 (15-de-Ac-T-2), 3′-OH-T-2, neosolaniol (NEO), T-2 triol, 3′-OH-HT-2, T-2 triol, Glucuronides: Gluc-3-T-2 | NO | Phase I metabolites: HT-2, 15-de-Ac-T-2, 3′-OH-T-2 and its two isomers, NEO or T-2 triol, 3′-OH-HT-2, Glucuronides: Gluc-3-T-2 | Two isomers of 3′-OH-T-2, 4 isomers of 3′-OH-HT-2 |
| HT-2 | Phase I metabolites: 4-de-Ac-NEO, 3′-OH-HT-2, 4′-OH-HT-2 7-OH-HT-2 and its isomer, 10-OH-HT-2, Glucuronides: Gluc-3-HT-2, Gluc-4-HT-2, Gluc-3-4-de-Ac-NEO | Gluc-4-HT-2, Gluc-3-4-de-Ac-NEO | Phase I metabolites: 4-de-Ac-NEO and its isomer, 3′-OH-HT-2, 4′-OH-HT-2 and its isomer Three OH-T-2 metabolites at 7 or 10 or 16-OH-HT-2 Two unknown metabolites Glucuronides: Gluc-3-HT-2 | 4-de-Ac-NEO isomer |
| 3-AcDON | Phase I metabolites: DON Glucuronides: Gluc-3-AcDON | NO | Phase I metabolites: DON Glucuronides: Gluc-3-AcDON | NO |
| 15-AcDON | Phase I metabolites: DON Glucuronides: Gluc-15-AcDON | NO | Phase I metabolites: DON Glucuronides: Gluc-15-AcDON | NO |
| FUS-X | Phase I metabolites: NIV | NO | Phase I metabolites: NIV Glucuronides: Gluc-FUS-X | Gluc-FUS-X |
| DON | Phase I metabolites: De-epoxy-DON (DOM-1) Glucuronides: 15-Gluc-DON, 3-Gluc-DON | NO | Phase I metabolites: NIV, DOM-1 and its two isomers, Glucuronides: 15-Gluc-DON, 3-Gluc-DON | NIV, isomers of DOM-1 |
| NIV | Phase I metabolites: De-epoxy-NIV (DENIV), Glucuronides: Gluc-3-NIV | NO | Phase I metabolites: DENIV and its two isomer Glucuronides: Two Gluc-NIVs | Gluc-NIV |
| AFB1 | Phase I metabolites: AFM1, AFQ1, AFBO, AFP1, AFL, AFB1-diol Glucuronides: NO | AFQ1 | Phase I metabolites: AFM1, AFBO, AFP1 and its isomer, AFL and its isomer, AFB1-diol and its isomer, ((H2)+(O)-AFB1 Glucuronides: NO | ((H2)+(O)-AFB1 |
| AFB2 | Phase I metabolites: AFM2, AFQ2, AB2A, AFP2 Glucuronides: NO | AFP2 | Phase I metabolites: AFM2, AFQ2 and AFB2A Glucuronides: NO | NO |
| AFG1 | Phase I metabolites: AFGM1 Glucuronides: NO | NO | Phase I metabolites: AFGM1 Glucuronides: NO | NO |
| AFG2 | Phase I metabolites: AFGM2, AFG2A Glucuronides: NO | NO | Phase I metabolites: AFGM2, AFG2A Glucuronides: NO | NO |
| ZEN | Phase I metabolites: (-(H2) +(O))-ZEN, (+(O))-ZEN, (+(H2)+ (O))-ZEN Glucuronides: Gluc-16-ZEN, Gluc-14-ZEN, Gluc-(+O)-ZEN, 2xGluc-ZEN | NO | Phase I metabolites: (-(H2) +(O))-ZEN, (+(O))-ZEN, (+(H2)+ (O))-ZEN Glucuronides: Gluc-16-ZEN, Gluc-14-ZEN, Gluc-(+O)-ZEN, 2xGluc-ZEN | NO |
| α-ZOL | Phase I metabolites: (-(H4)+(O))- α-ZOL (-(H2)+(O))- α-ZOL (+O)- α-ZOL Glucuronides: Gluc-16- α-ZOL, Gluc-14- α-ZOL, Gluc-7- α-ZOL | NO | Phase I metabolites: (-(H4)+(O))- α-ZOL (-(H2)+(O))- α-ZOL (+O)- α-ZOL Glucuronides: Gluc-16- α-ZOL, Gluc-14- α-ZOL, Gluc-7- α-ZOL, Gluc-(+O)- α-ZOL, 2 × Gluc- α-ZOL | Gluc-(+O)- α-ZOL, (2 × Gluc)- α-ZOL |
| β-ZOL | Phase I metabolites: NO Glucuronides: Gluc-16- β-ZOL, Gluc-14- β-ZOL, Gluc-7- β-ZOL, | NO | Phase I metabolites: (-(H4)+(O))- β-ZOL (-(H2)+(O))- β-ZOL (+O)- β-ZOL Glucuronides: Gluc-16- β-ZOL, Gluc-14- β-ZOL, Gluc-7- β-ZOL, Gluc-(+O)- β-ZOL, 2 × Gluc- β-ZOL | (-(H4)+(O))- β-ZOL (-(H2)+(O))- β-ZOL (+O)- β-ZOL Gluc-(+O)- β-ZOL, (2 × Gluc)- β-ZOL |
| ZAN | Phase I metabolites: NO Glucuronides: Gluc-16- ZAN, Gluc-14-ZAN | NO | Phase I metabolites: (-(H2) +(O))-ZAN, (+(O))-ZAN, (+(H2)+ (O))-ZAN, (+(O2))-ZAN, (-(H2) +(O2))-ZAN Glucuronides: Gluc-16-ZAN, Gluc-14-ZAN, Gluc-(+O)- ZAN, 2xGluc- ZAN, Gluc-(+(H2)+(O))-ZAN, 2 × Gluc-(+O)-ZAN, 2 × Gluc-(+(H2)+(O))-ZAN | Gluc-(+O)- ZAN, (2 × Gluc)- ZAN, Gluc-(+(H2)+(O))-ZAN, (2 × Gluc)-(+O)-ZAN, (2 × Gluc)-(+(H2)+(O))-ZAN |
| α-ZAL | Phase I metabolites: (-(H4)+(O))- α-ZAL (-(H2)+(O))- α-ZAL (+O)- α-ZAL (-(H4) +(O2))- α-ZAL (-(H2 )+(O2))- α-ZAL Glucuronides: Gluc-16- α-ZAL, Gluc-14- α-ZAL, Gluc-7- α-ZAL, | NO | Phase I metabolites: (-(H4)+(O))- α-ZAL (-(H2)+(O))- α-ZAL (+O)- α-ZAL (-(H4) +(O2))- α-ZAL (-(H2 )+(O2))- α-ZAL Glucuronides: Gluc-16- α-ZAL, Gluc-14- α-ZAL, Gluc-7- α-ZAL, Gluc-(+O)- α-ZAL 2 × Gluc-α-ZAL | Gluc-(+O)- α-ZAL (2 × Gluc)-α-ZAL |
| β-ZAL | Phase I metabolites: NO Glucuronides: Gluc-16-β-ZAL, Gluc-14-β-ZAL, Gluc-7-β-ZAL | NO | Phase I metabolites: (-(H4)+(O))- β-ZAL (-(H2)+(O))- β-ZAL (+O)- β-ZAL (-(H4) +(O2))- β-ZAL (-(H2 )+(O2))- β-ZAL Glucuronides: Gluc-16-β-ZAL, Gluc-14-β-ZAL, Gluc-7-β-ZAL, Gluc-(+O)- β-ZAL, 2xGluc-β-ZAL | (−(H4)+(O))- β-ZAL (−(H2)+(O))- β-ZAL (+O)- β-ZAL (−(H4) +(O2))- β-ZAL (−(H2 )+(O2))- β-ZALGluc-(+O)- β-ZAL, (2xGluc)-β-ZAL |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Slobodchikova, I.; Sivakumar, R.; Rahman, M.S.; Vuckovic, D. Characterization of Phase I and Glucuronide Phase II Metabolites of 17 Mycotoxins Using Liquid Chromatography—High-Resolution Mass Spectrometry. Toxins 2019, 11, 433. https://doi.org/10.3390/toxins11080433
Slobodchikova I, Sivakumar R, Rahman MS, Vuckovic D. Characterization of Phase I and Glucuronide Phase II Metabolites of 17 Mycotoxins Using Liquid Chromatography—High-Resolution Mass Spectrometry. Toxins. 2019; 11(8):433. https://doi.org/10.3390/toxins11080433
Chicago/Turabian StyleSlobodchikova, Irina, Reajean Sivakumar, Md Samiur Rahman, and Dajana Vuckovic. 2019. "Characterization of Phase I and Glucuronide Phase II Metabolites of 17 Mycotoxins Using Liquid Chromatography—High-Resolution Mass Spectrometry" Toxins 11, no. 8: 433. https://doi.org/10.3390/toxins11080433
APA StyleSlobodchikova, I., Sivakumar, R., Rahman, M. S., & Vuckovic, D. (2019). Characterization of Phase I and Glucuronide Phase II Metabolites of 17 Mycotoxins Using Liquid Chromatography—High-Resolution Mass Spectrometry. Toxins, 11(8), 433. https://doi.org/10.3390/toxins11080433