Isolation of a Novel Microcystin-Degrading Bacterium and the Evolutionary Origin of mlr Gene Cluster


Abstract
:1. Introduction
2. Results and Discussion
2.1. Isolation and Degradation Activity of MC-Degrading Bacterium
2.2. Whole mlr Gene Cluster in Strain X20
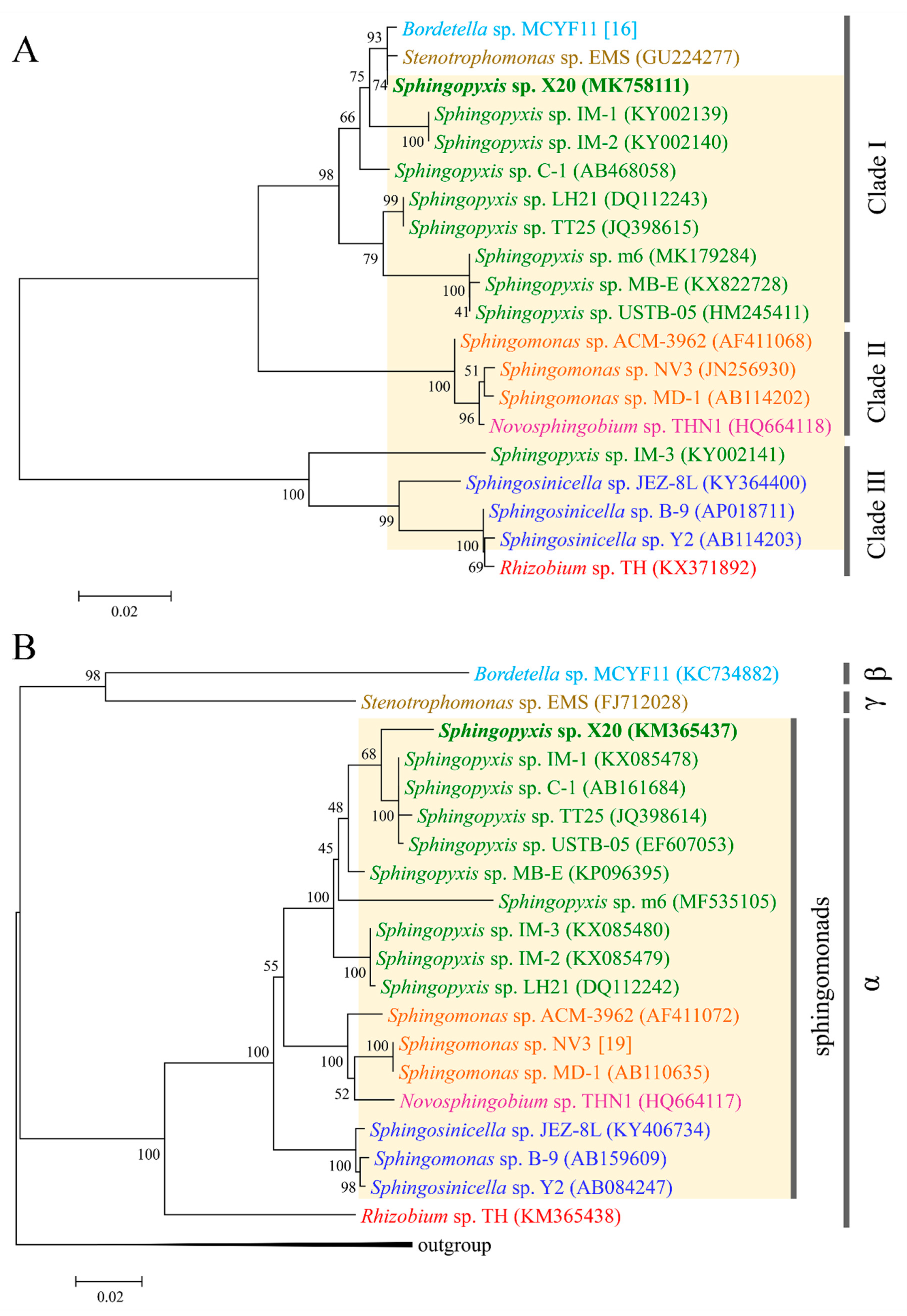
2.3. Evolutionary Origin of mlr Gene Cluster
3. Conclusions
4. Materials and Methods
4.1. Materials and Reagents
4.2. Isolation and Identification of MC-Degrading Bacterium
4.3. MCLR Degradation Experiments
4.4. Sequencing of the mlr Gene Cluster
4.5. Heterogeneous Expression of the mlrA Gene
4.6. Phylogenetic Analyses
4.7. Genomic Island Analyses
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Huisman, J.; Codd, G.A.; Paerl, H.W.; Ibelings, B.W.; Verspagen, J.M.H.; Visser, P.M. Cyanobacterial blooms. Nat. Rev. Microbiol. 2018, 16, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, R.P.; Sinha, R.P.; Incharoensakdi, A. The cyanotoxin-microcystins: Current overview. Rev. Environ. Sci. Biotechnol. 2014, 13, 215–249. [Google Scholar] [CrossRef]
- Li, J.; Li, R.; Li, J. Current research scenario for microcystins biodegradation—A review on fundamental knowledge, application prospects and challenges. Sci. Total Environ. 2017, 595, 615–632. [Google Scholar] [CrossRef]
- WHO. Guidelines for Drinking-Water Quality—Second Edition. In Addendum to Volume 2: Health Criteria and Other Supporting Information; World Health Organization: Geneva, Switzerland, 1998. [Google Scholar]
- Hall, T.; Hart, J.; Croll, B.; Gregory, R. Laboratory-scale investigations of algal toxin removal by water treatment. Water Environ. J. 2000, 14, 143–149. [Google Scholar] [CrossRef]
- Ho, L.; Sawade, E.; Newcombe, G. Biological treatment options for cyanobacteria metabolite removal—A review. Water Res. 2012, 46, 1536–1548. [Google Scholar] [CrossRef]
- Bourne, D.G.; Riddles, P.; Jones, G.J.; Smith, W.; Blakeley, R.L. Characterisation of a gene cluster involved in bacterial degradation of the cyanobacterial toxin microcystin LR. Environ. Toxicol. 2001, 16, 523–534. [Google Scholar] [CrossRef]
- Bourne, D.G.; Jones, G.J.; Blakeley, R.L.; Jones, A.; Negri, A.P.; Riddles, P. Enzymatic pathway for the bacterial degradation of the cyanobacterial cyclic peptide toxin microcystin LR. Appl. Environ. Microbiol. 1996, 62, 4086–4094. [Google Scholar] [PubMed]
- Shimizu, K.; Maseda, H.; Okano, K.; Kurashima, T.; Kawauchi, Y.; Xue, Q.; Utsumi, M.; Zhang, Z.; Sugiura, N. Enzymatic pathway for biodegrading microcystin LR in Sphingopyxis sp. C-1. J. Biosci. Bioeng. 2012, 114, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Dziga, D.; Wasylewski, M.; Szetela, A.; Bochenska, O.; Wladyka, B. Verification of the Role of MlrC in Microcystin Biodegradation by Studies Using a Heterologously Expressed Enzyme. Chem. Res. Toxicol. 2012, 25, 1192–1194. [Google Scholar] [CrossRef]
- Dziga, D.; Zielinska, G.; Wladyka, B.; Bochenska, O.; Maksylewicz, A.; Strzalka, W.; Meriluoto, J. Characterization of Enzymatic Activity of MlrB and MlrC Proteins Involved in Bacterial Degradation of Cyanotoxins Microcystins. Toxins 2016, 8, 76. [Google Scholar] [CrossRef]
- Fontanillo, M.; Köhn, M. Microcystins: Synthesis and structure–activity relationship studies toward PP1 and PP2A. Bioorg. Med. Chem. 2018, 26, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Shen, Y.; Chen, X.; Hu, Y.O.O.; Xiang, H.; Tao, J.; Ling, Y. Biodegradation mechanism of microcystin-LR by a novel isolate of Rhizobium sp. TH and the evolutionary origin of the mlrA gene. Int. Biodeterior. Biodegrad. 2016, 115, 17–25. [Google Scholar] [CrossRef]
- Kormas, K.A.; Lymperopoulou, D.S. Cyanobacterial toxin degrading bacteria: Who are they? Biomed. Res. Int. 2013. [Google Scholar] [CrossRef] [PubMed]
- Thees, A.; Atari, E.; Birbeck, J.; Westrick, J.A.; Huntley, J.F. Isolation and characterization of Lake Erie bacteria that degrade the cyanobacterial microcystin toxin MC-LR. J. Great Lakes Res. 2019, 45, 138–149. [Google Scholar] [CrossRef]
- Krishnan, A.; Zhang, Y.Q.; Mou, X.Z. Isolation and Characterization of Microcystin-Degrading Bacteria from Lake Erie. Bull. Environ. Contam Toxicol. 2018, 101, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhou, Y.; Sun, R.; Wei, H.; Li, Y.; Yin, L.; Pu, Y. Biodegradation of microcystin-LR and-RR by a novel microcystin-degrading bacterium isolated from Lake Taihu. Biodegradation 2014, 25, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Maghsoudi, E.; Fortin, N.; Greer, C.; Maynard, C.; Page, A.; Vo Duy, S.; Sauve, S.; Prevost, M.; Dorner, S. Cyanotoxin degradation activity and mlr gene expression profiles of a Sphingopyxis sp. isolated from Lake Champlain, Canada. Environ. Sci. Process. Impacts 2016, 18, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Somdee, T.; Thunders, M.; Ruck, J.; Lys, I.; Allison, M.; Page, R. Degradation of [Dha(7)]MC-LR by a Microcystin Degrading Bacterium Isolated from Lake Rotoiti, New Zealand. ISRN Microbiol. 2013, 2013, 596429. [Google Scholar] [CrossRef]
- Jiang, Y.; Shao, J.; Wu, X.; Xu, Y.; Li, R. Active and silent members in the mlr gene cluster of a microcystin-degrading bacterium isolated from Lake Taihu, China. FEMS Microbiol. Lett. 2011, 322, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Hoefel, D.; Saint, C.P.; Newcombe, G. Isolation and identification of a novel microcystin-degrading bacterium from a biological sand filter. Water Res. 2007, 41, 4685–4695. [Google Scholar] [CrossRef]
- Zhang, M.; Pan, G.; Yan, H. Microbial biodegradation of microcystin-RR by bacterium Sphingopyxis sp. USTB-05. J. Environ. Sci. 2010, 22, 168–175. [Google Scholar] [CrossRef]
- Saito, T.; Okano, K.; Park, H.D.; Itayama, T.; Inamori, Y.; Neilan, B.A.; Burns, B.P.; Sugiura, N. Detection and sequencing of the microcystin LR-degrading gene, mlrA, from new bacteria isolated from Japanese lakes. FEMS Microbiol. Lett. 2003, 229, 271–276. [Google Scholar] [CrossRef]
- Zhu, L.; Wu, Y.; Song, L.; Gan, N. Ecological dynamics of toxic Microcystis spp. and microcystin-degrading bacteria in Dianchi Lake, China. Appl. Environ. Microbiol. 2014, 80, 1874–1881. [Google Scholar] [CrossRef]
- Jones, G.J.; Bourne, D.G.; Blakeley, R.L.; Doelle, H. Degradation of the cyanobacterial hepatotoxin microcystin by aquatic bacteria. Nat. Toxins 1994, 2, 228–235. [Google Scholar] [CrossRef]
- Tsuji, K.; Asakawa, M.; Anzai, Y.; Sumino, T.; Harada, K.-I. Degradation of microcystins using immobilized microorganism isolated in an eutrophic lake. Chemosphere 2006, 65, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-D.; Sasaki, Y.; Maruyama, T.; Yanagisawa, E.; Hiraishi, A.; Kato, K. degradation of the cyanobacterial hepatotoxin microcystin by a new bacterium isolated from a hypertrophic lake. Environ. Toxicol. 2001, 16, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A. Molecular characteristics of xenobiotic-degrading sphingomonads. Appl. Microbiol. Biotechnol. 2009, 81, 793–811. [Google Scholar] [CrossRef]
- Lezcano, M.Á.; Velázquez, D.; Quesada, A.; El-Shehawy, R. Diversity and temporal shifts of the bacterial community associated with a toxic cyanobacterial bloom: An interplay between microcystin producers and degraders. Water Res. 2017, 125, 52–61. [Google Scholar] [CrossRef]
- Wang, R.P.; Li, J.M.; Jiang, Y.G.; Lu, Z.J.; Li, R.H.; Li, J. Heterologous expression of mlrA gene originated from Novosphingobium sp. THN1 to degrade microcystin-RR and identify the first step involved in degradation pathway. Chemosphere 2017, 184, 159–167. [Google Scholar] [CrossRef]
- Chen, J.; Hu, L.B.; Zhou, W.; Yan, S.H.; Yang, J.D.; Xue, Y.F.; Shi, Z.Q. Degradation of Microcystin-LR and RR by a Stenotrophomonas sp. Strain EMS Isolated from Lake Taihu, China. Int. J. Mol. Sci. 2010, 11, 896–911. [Google Scholar] [CrossRef]
- Jin, H.Y.; Hiraoka, Y.; Okuma, Y.; Hashimoto, E.H.; Kurita, M.; Anas, A.R.J.; Uemura, H.; Tsuji, K.; Harada, K.I. Microbial Degradation of Amino Acid-Containing Compounds Using the Microcystin-Degrading Bacterial Strain B-9. Mar. Drugs 2018, 16, 50. [Google Scholar] [CrossRef] [PubMed]
- Okano, K.; Shimizu, K.; Maseda, H.; Kawauchi, Y.; Utsumi, M.; Itayama, T.; Zhang, Z.; Sugiura, N. Whole-genome sequence of the microcystin-degrading bacterium Sphingopyxis sp. strain C-1. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, X.; Yang, L.; Xiao, B.; Wu, X.; Wang, J.; Wan, H. An effective pathway for the removal of microcystin LR via anoxic biodegradation in lake sediments. Water Res. 2010, 44, 1884–1892. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Sequence (5′–3′) | Purpose | References |
|---|---|---|---|---|
| mlrC-mlrA | mlrCf1 | TCCCCGAAACCGATTCTCCA | Partial mlr | [21] |
| MR | CTCCTCCCACAAATCAGGAC | [23] | ||
| mlrA-mlrD | MF | GACCCGATGTTCAAGATACT | Partial mlr | [23] |
| mlrDr1 | ACAGTGTTGCCGAGCTGCTCA | [21] | ||
| mlrD-mlrB | mlrDf1 | GCTGGCTGCGACGGAAATG | Partial mlr | [21] |
| mlrBr1 | CGTGCGGACTACTGTTGG | |||
| mlrB | mlrBf2 | ATGACTGCAACAAAGCTTTT | Partial mlr | This study |
| mlrBr2 | TTATCCACGAACAACCCACC | |||
| mlrC | CR1 | CCCTGGCAGTACAATTGGGCTTTGA | Flanking region | This study |
| CR2 | CACAGGGCTTGCCGAGAATGTCA | |||
| CR3 | CGTCAGCGAAATTCGCGACCAGT | |||
| mlrB | BF1 | AGGTAGGTCAGGCAGATAGGTG | Flanking region | This study |
| BF2 | AAGATCAGGATGAGAACGGCCG | |||
| BF3 | AGATCAGCAAGTCCAAAGCCGC | |||
| mlrA | MlrAxf | GACGGATCCATGCGGGAGTTTGTCAAAC | Expression | This study |
| MlrAxr | TATAAGCTTCGCGTTCGCGCCGGACTTG | |||
| mlrE | mlrEf | TTCGGTAGACGGAACACA | GI verification | This study |
| mlrEr | ACACGGCATTGATCTGAAT | |||
| mlrF | mlrFf | GATGGAAGAGGTGATGGCAATT | GI verification | This study |
| mlrFr | AGGACGAATACTGGTGGTAGTC | |||
| GI1 | G1f | ACTCTGGACCAGCGGCTAA | GI verification | This study |
| G1r | CAAGCGGACTGACAAGTTCTG | |||
| GI2 | G2f | GCAACCGTCATCAGTGGATC | GI verification | This study |
| G2r | CCGCCGTAGTATTCGTGAATG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, L.; Zhang, X.; Chen, X.; Wang, K.; Shen, Y.; Li, D. Isolation of a Novel Microcystin-Degrading Bacterium and the Evolutionary Origin of mlr Gene Cluster. Toxins 2019, 11, 269. https://doi.org/10.3390/toxins11050269
Qin L, Zhang X, Chen X, Wang K, Shen Y, Li D. Isolation of a Novel Microcystin-Degrading Bacterium and the Evolutionary Origin of mlr Gene Cluster. Toxins. 2019; 11(5):269. https://doi.org/10.3390/toxins11050269
Chicago/Turabian StyleQin, Lian, Xiaoxing Zhang, Xiaoguo Chen, Ke Wang, Yitian Shen, and Dan Li. 2019. "Isolation of a Novel Microcystin-Degrading Bacterium and the Evolutionary Origin of mlr Gene Cluster" Toxins 11, no. 5: 269. https://doi.org/10.3390/toxins11050269
APA StyleQin, L., Zhang, X., Chen, X., Wang, K., Shen, Y., & Li, D. (2019). Isolation of a Novel Microcystin-Degrading Bacterium and the Evolutionary Origin of mlr Gene Cluster. Toxins, 11(5), 269. https://doi.org/10.3390/toxins11050269