Novel Regulation of Alpha-Toxin and the Phenol-Soluble Modulins by Peptidyl-Prolyl cis/trans Isomerase Enzymes in Staphylococcus aureus

 , ,
, ,
Abstract
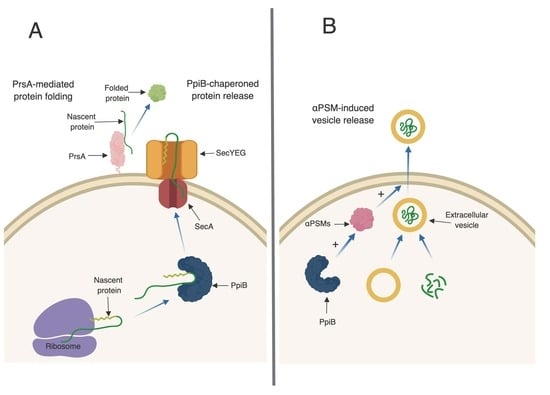
:
1. Introduction
2. Results
2.1. PpiB Is Required for Virulence in a Murine Abscess and Systemic Model of Infection
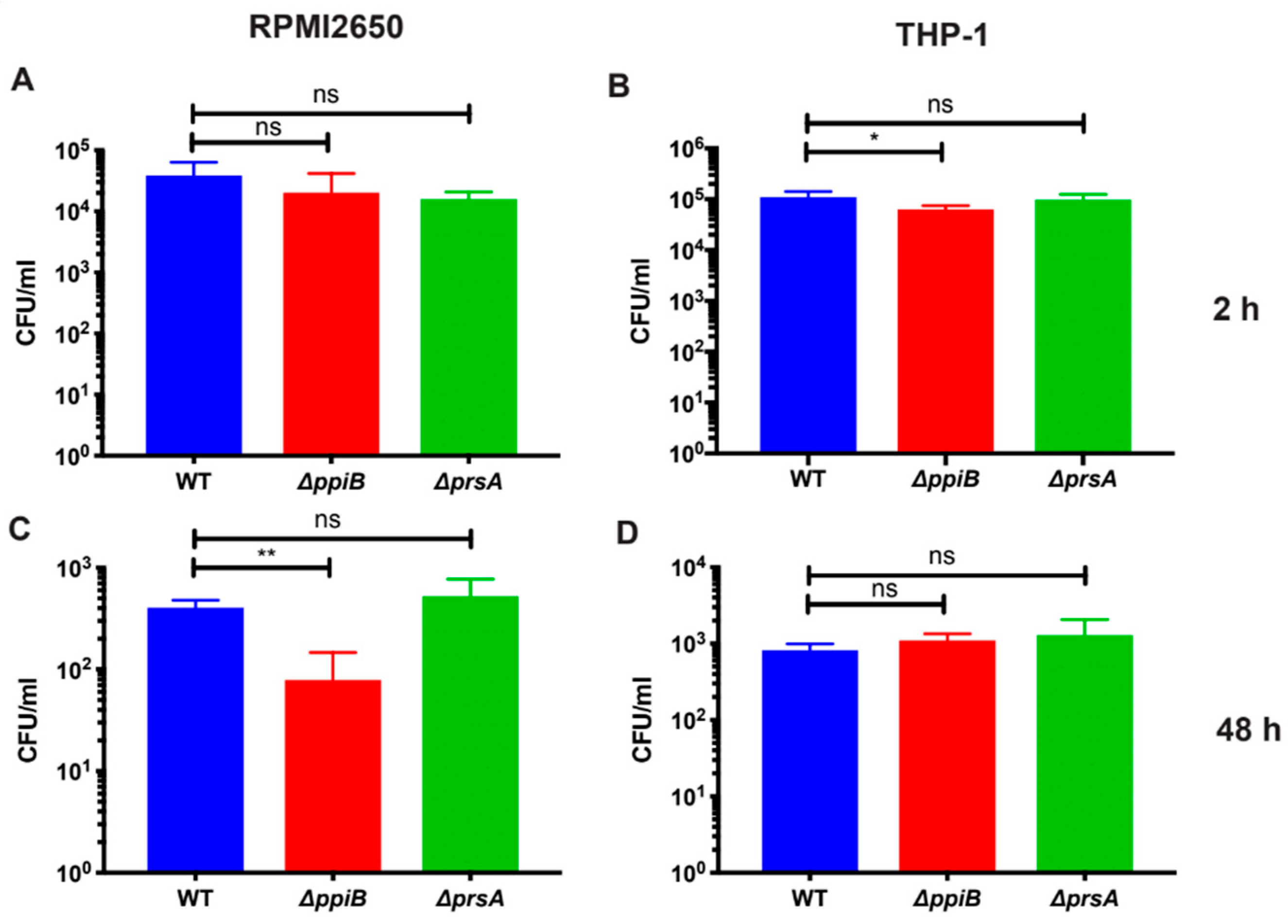
2.2. PpiB is Required for Survival inside Macrophages and Human Nasal-Epithelial Cells
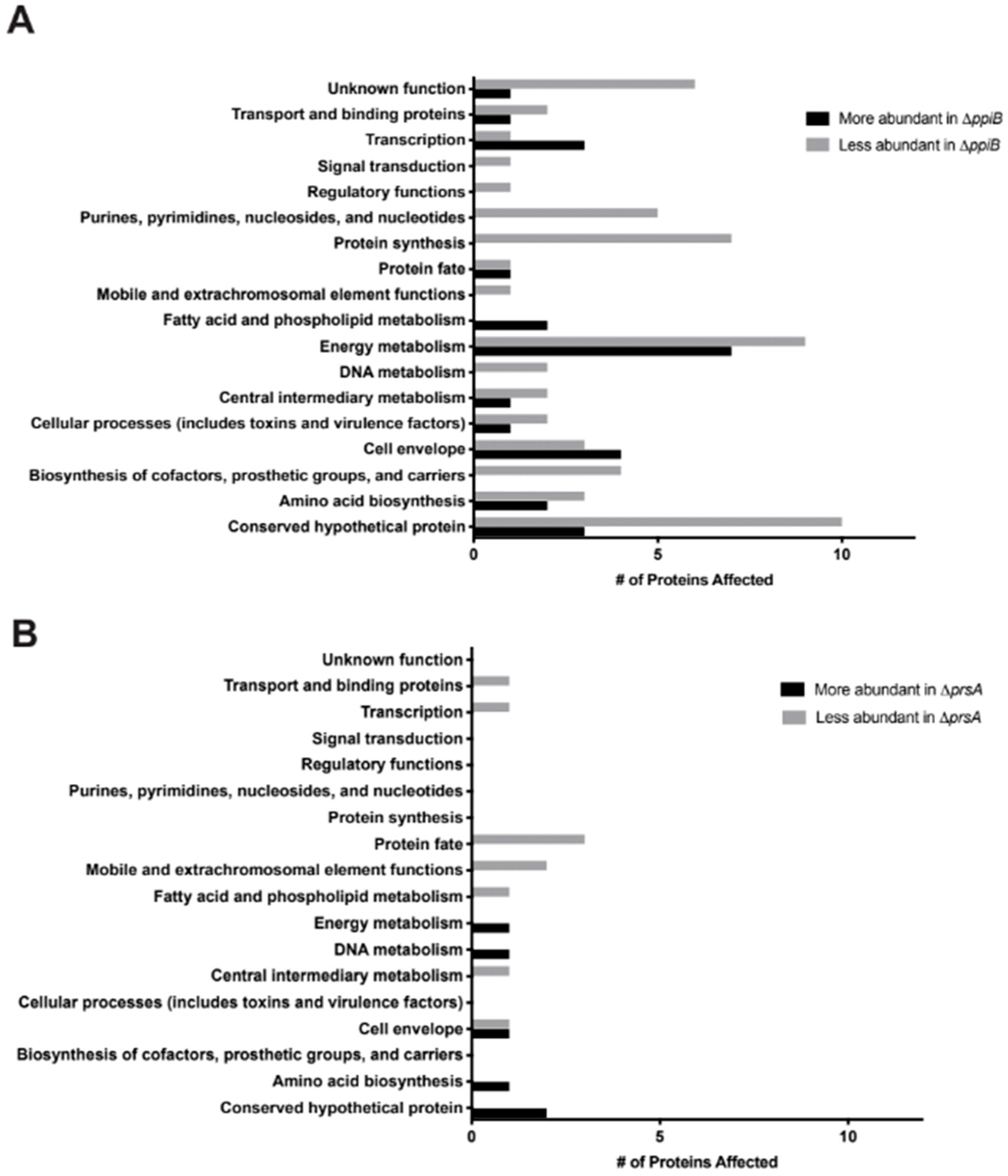
2.3. Exoproteome Analysis Reveals Greater Alterations in Secreted Protein Abundance in a ΔppiB Mutant Than a ΔprsA Mutant
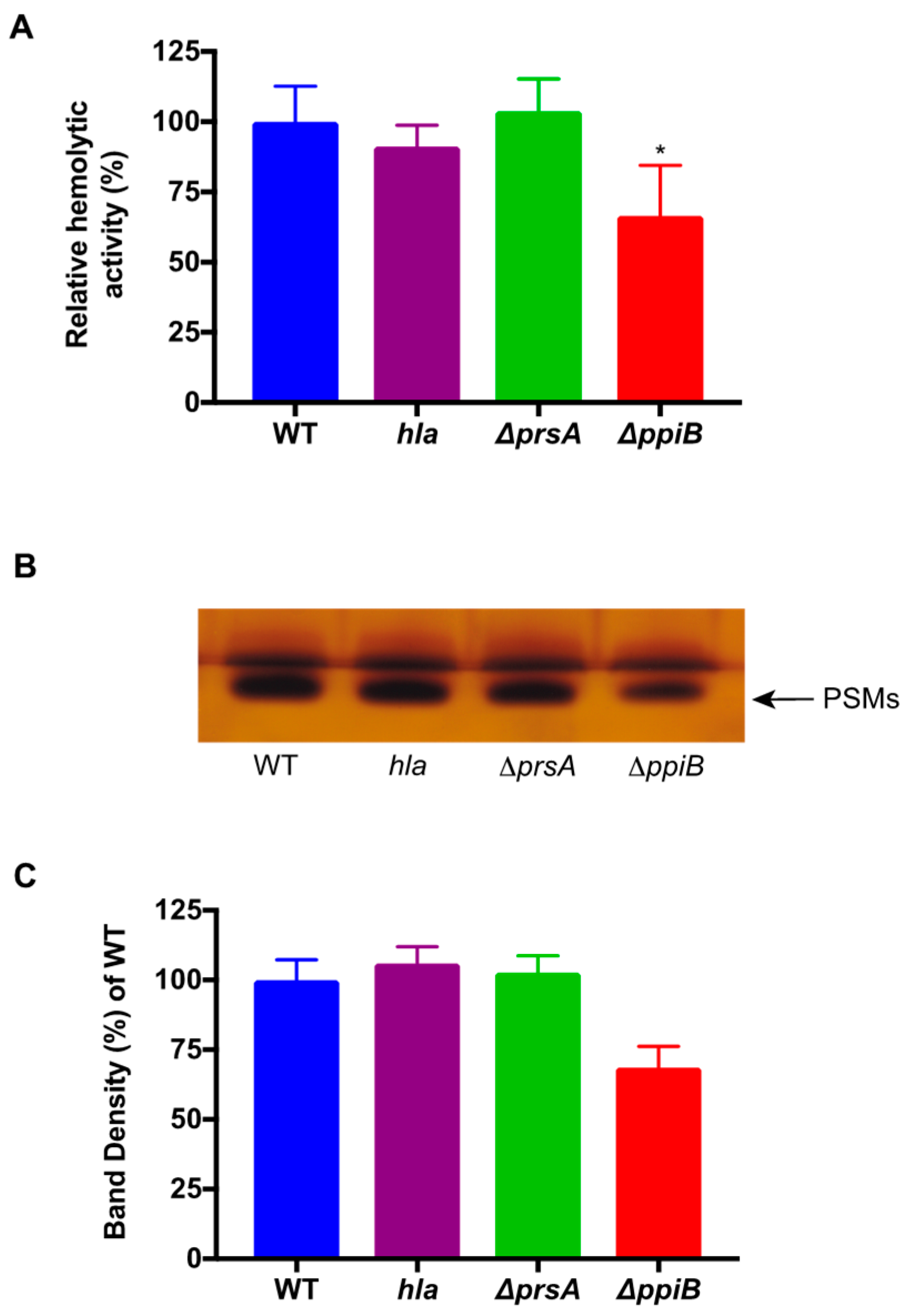
2.4. A ΔppiB Mutant Has Reduced PSM Production
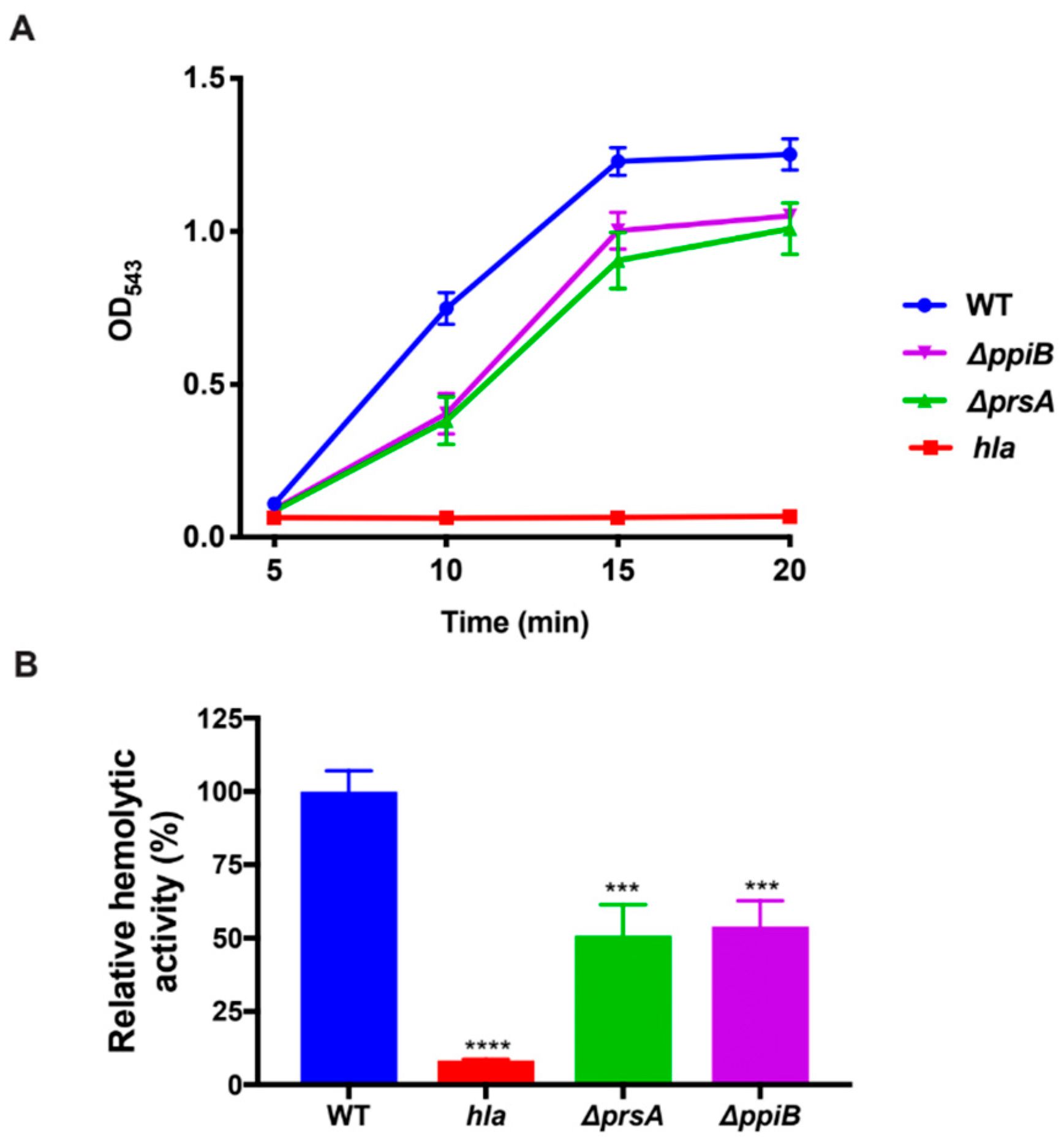
2.5. The ΔppiB and ΔprsA Mutants Both Display Reduced Hemolysis of Rabbit Erythrocytes
2.6. In Vivo Immunoprecipitation Identifies Greater Abundance of PpiB Target Proteins Than PrsA
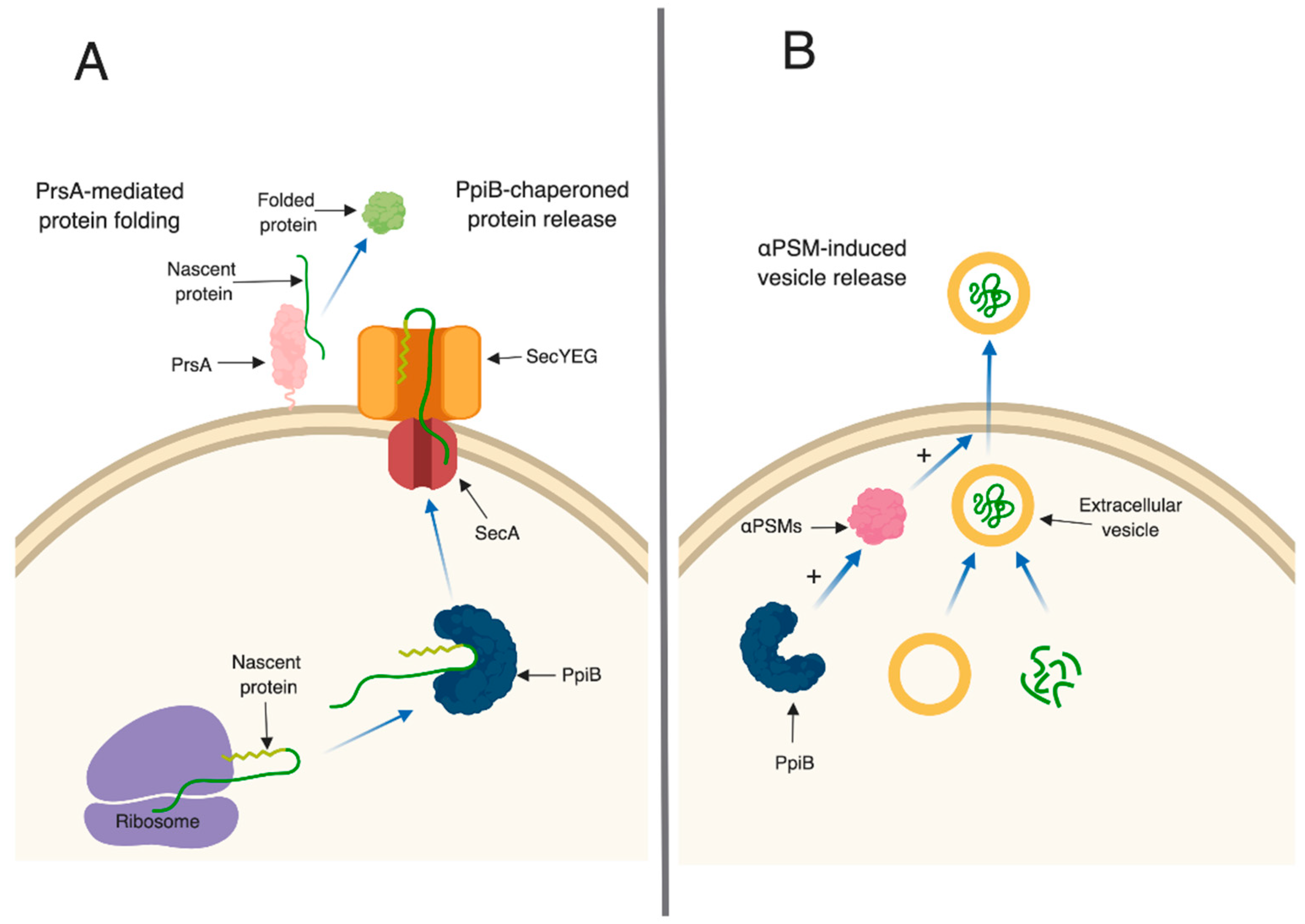
3. Discussion
4. Materials and Methods
4.1. Strains and Strain Construction
4.2. Bacterial Growth Conditions
4.3. Murine Abscess Model of Infection
4.4. Murine Systemic Model of Infection
4.5. Macrophage Infection and Cell Differentiation
4.6. Nasal Epithelial Cell Infection
4.7. Exoproteome Analysis
4.8. Butanol Extraction of PSMs and Densitometry Analysis
4.9. Human-Erythrocyte Hemolysis Assay
4.10. Rabbit Erythrocyte Hemolysis Assay
4.11. Protein Immunoprecipitation Assay
4.12. Reverse Transcriptase-Quantitative PCR (RT-qPCR)
4.13. Ethics Statement
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lu, T.; DeLeo, F.R. Pathogenesis of staphylococcus aureus in Humans. In Human Emerging and Re-emerging Infections; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 711–748. [Google Scholar]
- Otto, M. Staphylococcus aureus toxins. Curr. Opin. Microbiol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, A.; Diep, B.A.; Mai, T.T.; Vo, N.H.; Warrener, P.; Suzich, J.; Stover, C.K.; Sellman, B.R. Differential expression and roles of staphylococcus aureus virulence determinants during colonization and disease. MBio 2015, 6, e02272-14. [Google Scholar] [CrossRef] [PubMed]
- Mann, E.E.; Rice, K.C.; Boles, B.R.; Endres, J.L.; Ranjit, D.; Chandramohan, L.; Tsang, L.H.; Smeltzer, M.S.; Horswill, A.R.; Bayles, K.W. Modulation of eDNA release and degradation affects staphylococcus aureus biofilm maturation. PLoS ONE 2009, 4, e5822. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.J.; Henderson, B.; Nair, S.P. Staphylococcus aureus fibronectin binding proteins A and B possess a second fibronectin binding region that may have biological relevance to bone tissues. Calcif. Tissue Int. 2002, 70, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Bhakdil, S. Alpha-Toxin of Staphylococcus aureus. Microbiol. Mol. Biol. Rev. 1991, 55, 733–751. [Google Scholar]
- Joo, H.S.; Chatterjee, S.S.; Villaruz, A.E.; Dickey, S.W.; Tan, V.Y.; Chen, Y.; Sturdevant, D.E.; Ricklefs, S.M.; Otto, M. Mechanism of gene regulation by a staphylococcus aureus toxin. MBio 2016, 7, e01579-16. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.D.; Malachowa, N.; Whitney, A.R.; Braughton, K.R.; Gardner, D.J.; Long, D.; Wardenburg, J.B.; Schneewind, O.; Otto, M.; DeLeo, F.R. Comparative analysis of USA300 virulence determinants in a rabbit model of skin and soft tissue infection. J. Infect. Dis. 2011, 204, 937–941. [Google Scholar] [CrossRef]
- Wilke, G.A.; Wardenburg, J.B. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus—Hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. USA 2010, 107, 13473–13478. [Google Scholar] [CrossRef]
- Cheung, G.Y.C.; Joo, H.S.; Chatterjee, S.S.; Otto, M. Phenol-soluble modulins—Critical determinants of staphylococcal virulence. FEMS Microbiol. Rev. 2014, 38, 698–719. [Google Scholar] [CrossRef]
- Queck, S.Y.; Jameson-Lee, M.; Villaruz, A.E.; Bach, T.-H.L.; Khan, B.A.; Sturdevant, D.E.; Ricklefs, S.M.; Li, M.; Otto, M. RNAIII-independent target gene control by the agr quorum-sensing system: Insight into the evolution of virulence regulation in staphylococcus aureus. Mol. Cell 2008, 32, 150–158. [Google Scholar] [CrossRef]
- Berube, B.J.; Sampedro, G.R.; Otto, M.; Wardenburg, J.B. The psmα locus regulates production of staphylococcus aureus alpha-toxin during infection. Infect. Immun. 2014, 82, 3350–3358. [Google Scholar] [CrossRef] [PubMed]
- Kofron, J.L.; Kuzmic, P.; Kishore, V.; Colon-Bonilla, E.; Rich, D.H. Determination of kinetic constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay. Biochemistry 1991, 30, 6127–6134. [Google Scholar] [CrossRef] [PubMed]
- Unal, C.M.; Steinert, M. Microbial peptidyl-prolyl cis/trans isomerases (PPIases): Virulence factors and potential alternative drug targets. Microbiol. Mol. Biol. Rev. 2014, 78, 544–571. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, F.; Freitag, N.E. Listeria monocytogenes PrsA2 is required for virulence factor secretion and bacterial viability within the host cell cytosol. Infect. Immun. 2010, 78, 4944–4957. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, F.; Port, G.C.; Cao, M.; Freitag, N.E. The posttranslocation chaperone PrsA2 contributes to multiple facets of Listeria monocytogenes pathogenesis. Infect. Immun. 2009, 77, 2612–2623. [Google Scholar] [CrossRef]
- Reffuveille, F.; Connil, N.; Sanguinetti, M.; Posteraro, B.; Chevalier, S.; Auffray, Y.; Rince, A. Involvement of peptidylprolyl cis/trans isomerases in enterococcus faecalis virulence. Infect. Immun. 2012, 80, 1728–1735. [Google Scholar] [CrossRef] [PubMed]
- Roset, M.S.; Fernández, L.G.; Delvecchio, V.G.; Briones, G. Intracellularly induced cyclophilins play an important role in stress adaptation and virulence of brucella abortus. Infect. Immun. 2013, 81, 521–530. [Google Scholar] [CrossRef]
- Pandey, S.; Sharma, A.; Tripathi, D.; Kumar, A.; Khubaib, M.; Bhuwan, M.; Chaudhuri, T.K.; Hasnain, S.E.; Ehtesham, N.Z. Mycobacterium tuberculosis Peptidyl-Prolyl Isomerases Also Exhibit Chaperone like Activity In-Vitro and In-Vivo. PLoS ONE 2016, 11, e0150288. [Google Scholar] [CrossRef]
- Skagia, A.; Zografou, C.; Vezyri, E.; Venieraki, A.; Katinakis, P.; Dimou, M. Cyclophilin PpiB is involved in motility and biofilm formation via its functional association with certain proteins. Genes Cells 2016, 21, 833–851. [Google Scholar] [CrossRef] [Green Version]
- Keogh, R.A.; Zapf, R.L.; Wiemels, R.E.; Wittekind, M.A.; Carroll, R.K. The intracellular cyclophilin PpiB contributes to the virulence of staphylococcus aureus independently of Its peptidyl-prolyl cis/trans isomerase activity. Infect. Immun. 2018, 86, 1–13. [Google Scholar] [CrossRef]
- Lin, M.-H.; Li, C.-C.; Shu, J.-C.; Chu, H.-W.; Liu, C.-C.; Wu, C.-C. Exoproteome profiling reveals the involvement of the foldase PrsA in the cell surface properties and pathogenesis of staphylococcus aureus. Proteomics 2018, 18, 1700195. [Google Scholar] [CrossRef] [PubMed]
- Jousselin, A.; Manzano, C.; Biette, A.; Reed, P.; Pinho, M.G.; Rosato, A.E.; Kelley, W.L.; Renzoni, A. The Staphylococcus aureus Chaperone PrsA Is a New Auxiliary Factor of Oxacillin Resistance Affecting Penicillin-Binding Protein 2A. Antimicrob. Agents Chemother. 2016, 60, 1656–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiemels, R.E.; Cech, S.M.; Meyer, N.M.; Burke, C.A.; Weiss, A.; Parks, A.R.; Shaw, L.N.; Carroll, R.K. An intracellular peptidyl-prolyl cis/trans isomerase is required for folding and activity of the Staphylococcus aureus secreted virulence factor nuclease. J. Bacteriol. 2017, 199, e00453-16. [Google Scholar] [CrossRef] [PubMed]
- Malachowa, N.; Kobayashi, S.D.; Braughton, K.R.; DeLeo, F.R. Mouse Model of Staphylococcus Aureus Skin Infection; Humana Press: Totowa, NJ, USA, 2013; pp. 109–116. [Google Scholar]
- Alonzo, F., III; Benson, M.A.; Chen, J.; Novick, R.P.; Shopsin, B.; Torres, V.J. Staphylococcus aureus leucocidin ED contributes to systemic infection by targeting neutrophils and promoting bacterial growth in vivo. Mol. Microbiol. 2012, 83, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Thompson, C.D.; Weidenmaier, C.; Lee, J.C. Release of staphylococcus aureus extracellular vesicles and their application as a vaccine platform. Nat. Commun. 2018, 9, 1379. [Google Scholar] [CrossRef] [PubMed]
- Berube, B.J.; Wardenburg, J.B. Staphylococcus aureus α-toxin: Nearly a century of intrigue. Toxins 2013, 5, 1140–1166. [Google Scholar] [CrossRef] [PubMed]
- Otto, H.-S.J. The isolation and Analysis of Phenol-Soluble Modulins of Staphylococcus Epidermidis; Humana Press: Totowa, NJ, USA, 2014; pp. 93–100. [Google Scholar]
- Schallenberger, M.A.; Niessen, S.; Shao, C.; Fowler, B.J.; Romesberg, F.E. Type I signal peptidase and protein secretion in staphylococcus aureus. J. Bacteriol. 2012, 194, 2677–2686. [Google Scholar] [CrossRef] [PubMed]
- Antelmann, H.; Darmon, E.; Noone, D.; Veening, J.W.; Westers, H.; Bron, S.; Kuipers, O.P.; Devine, K.M.; Hecker, M.; Van Dijl, J.M. The extracellular proteome of Bacillus subtilis under secretion stress conditions. Mol. Microbiol. 2003, 49, 143–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigoulay, C.; Entenza, J.M.; Halpern, D.; Widmer, E.; Moreillon, P.; Poquet, I.; Gruss, A. Comparative analysis of the roles of HtrA-like surface proteases in two virulent staphylococcus aureus strains. Infect. Immun. 2005, 73, 563–572. [Google Scholar] [CrossRef]
- McCallum, N.; Spehar, G.; Bischoff, M.; Berger-Bächi, B. Strain dependence of the cell wall-damage induced stimulon in staphylococcus aureus. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2006, 1760, 1475–1481. [Google Scholar] [CrossRef]
- Schneewind, O.; Missiakas, D. Sec-secretion and sortase-mediated anchoring of proteins in Gram-positive bacteria. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2014, 1843, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Pugsley, A.P. The complete general secretory pathway in gram-negative bacteria. Microbiol. Rev. 1993, 57, 50–108. [Google Scholar] [PubMed]
- Jousselin, A.; Renzoni, A.; Andrey, D.O.; Monod, A.; Lew, D.P.; Kelley, W.L. The posttranslocational chaperone lipoprotein PrsA Is Involved in both glycopeptide and oxacillin resistance in staphylococcus aureus. Antimicrob. Agents Chemother. 2012, 56, 3629–3640. [Google Scholar] [CrossRef] [PubMed]
- Skagia, A.; Vezyri, E.; Sigala, M.; Kokkinou, A.; Karpusas, M.; Venieraki, A.; Katinakis, P.; Dimou, M. Structural and functional analysis of cyclophilin PpiB mutants supports an in vivo function not limited to prolyl isomerization activity. Genes Cells 2017, 22, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Göthel, S.F.; Marahiel, M.A. Peptidyl-prolyl cis-trans isomerases, a superfamily of ubiquitous folding catalysts. Cells Mol. Life Sci. 1999, 55, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Herbert, S.; Ziebandt, A.K.; Ohlsen, K.; Schäfer, T.; Hecker, M.; Albrecht, D.; Novick, R.; Götz, F. Repair of global regulators in staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect. Immun. 2010, 78, 2877–2889. [Google Scholar] [CrossRef] [PubMed]
- Buchner, J. Supervising the fold: Functional principles of molecular chaperones. FASEB J. 1996, 10, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Bose, J.L. Genetic manipulation of staphylococci. In Methods in Molecular Biology; Humana Press: Clifton, NJ, USA, 2014; pp. 101–111. [Google Scholar]
- Bose, J.L.; Fey, P.D.; Bayles, K.W. Genetic tools to enhance the study of gene function and regulation in staphylococcus aureus. Appl. Environ. Microbiol. 2013, 79, 2218–2224. [Google Scholar] [CrossRef]
- Kreiswirth, B.N.; Löfdahl, S.; Betley, M.J.; O’Reilly, M.; Schlievert, P.M.; Bergdoll, M.S.; Novick, R.P. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 1983, 305, 709. [Google Scholar] [CrossRef]
- Highlander, S.K.; Hultén, K.G.; Qin, X.; Jiang, H.; Yerrapragada, S.; Mason, E.O.; Shang, Y.; Williams, T.M.; Fortunov, R.M.; Liu, Y.; et al. Subtle genetic changes enhance virulence of methicillin resistant and sensitive Staphylococcus aureus. BMC Microbiol. 2007, 7, 1–14. [Google Scholar] [CrossRef]
- Fey, P.D.; Endres, J.L.; Yajjala, V.K.; Widhelm, T.J.; Boissy, R.J.; Bose, J.L.; Bayles, K.W. A genetic resource for rapid and comprehensive phenotype screening of nonessential staphylococcus aureus genes. MBio 2013, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Boles, B.R.; Thoende, M.; Roth, A.J.; Horswill, A.R. Identification of genes involved in polysaccharide- independent staphylococcus aureus biofilm formation. PLoS ONE 2010, 5, e10146. [Google Scholar] [CrossRef] [PubMed]
- Zapf, R.L.; Wiemels, R.E.; Keogh, R.A.; Holzschu, D.L.; Howell, K.M.; Trzeciak, E.; Caillet, A.R.; King, K.A.; Selhorst, S.A.; Naldrett, M.J.; et al. The small RNA Teg41 regulates expression of the alpha phenol-soluble modulins and Is required for Virulence in staphylococcus aureus. MBio 2019, 10, e02484-18. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.A.; Yasbin, R.E.; Young, F.E. New shuttle vectors for bacillus subtilis and escherichia coli which allow rapid detection of inserted fragments. Gene 1984, 29, 21–26. [Google Scholar] [CrossRef]
- Spaan, A.N.; Reyes-Robles, T.; Badiou, C.; Cochet, S.; Boguslawski, K.M.; Yoong, P.; Day, C.J.; De Haas, C.J.C.; Van Kessel, K.P.M.; Vandenesch, F.; et al. Staphylococcus aureus targets the duffy antigen receptor for chemokines (DARC) to lyse erythrocytes. Cell Host Microbe 2015, 18, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Carroll, R.K.; Rivera, F.E.; Cavaco, C.K.; Johnson, G.M.; Martin, D.; Shaw, L.N. The lone S41 family C-terminal processing protease in Staphylococcus aureus is localized to the cell wall and contributes to virulence. Microbiology 2014, 160, 1737–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, A.; Mann, M.; Aebersold, R.; Goodlett, D.R.; Link, A.J.; Eng, J.; Schieltz, D.M.; Carmack, E. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Designation | Protein Name | Fold Change 1 | Functional Grouping |
|---|---|---|---|
| SAUSA300_1533 | YdfA | 25.51 | Conserved hypothetical protein |
| SAUSA300_0062 | ArcB | 23.74 | Amino acid biosynthesis |
| SAUSA300_0962 | QoxB | 18.74 | Energy metabolism |
| SAUSA300_0226 | FadB | 13.36 | Fatty acid and phospholipid metabolism |
| SAUSA300_2581 | SasA | 7.36 | Cell envelope |
| SAUSA300_1305 | OdhB | 4.93 | Energy metabolism |
| SAUSA300_0912 | FabI | 4.78 | Fatty acid and phospholipid metabolism |
| SAUSA300_1594 | YajC | 4.71 | Protein fate |
| SAUSA300_2061 | AtpH | 3.70 | Energy metabolism |
| SAUSA300_2060 | AtpA | 3.44 | Energy metabolism |
| SAUSA300_0547 | SdrD | 3.37 | Cell envelope |
| SAUSA300_2143 | 3.32 | Conserved hypothetical protein | |
| SAUSA300_2178 | RpoA | 3.23 | Transcription |
| SAUSA300_2058 | AtpD | 3.13 | Energy metabolism |
| SAUSA300_0527 | RpoB | 2.87 | Transcription |
| SAUSA300_2059 | AtpG | 2.80 | Energy metabolism |
| SAUSA300_0528 | RpoC | 2.63 | Transcription |
| SAUSA300_0194 | MurP | 2.49 | Cellular processes (includes toxins and virulence factors) |
| SAUSA300_1565 | 2.40 | Central intermediary metabolism | |
| SAUSA300_1685 | 2.28 | Conserved hypothetical protein | |
| SAUSA300_2453 | NcaC | 2.23 | Transport and binding proteins |
| SAUSA300_2440 | FnbB | 2.17 | Cell envelope |
| SAUSA300_0724 | 2.10 | Cell envelope | |
| SAUSA300_0514 | CysE | 2.08 | Amino acid biosynthesis |
| SAUSA300_0963 | QoxA | 2.05 | Energy metabolism |
| SAUSA300_2573 | IsaB | 2.00 | Unknown function |
| SAUSA300_1881 | GatA | 0.50 | Protein synthesis |
| SAUSA300_0691 | SaeR | 0.49 | Regulatory functions |
| SAUSA300_2469 | SdaAA | 0.49 | Energy metabolism |
| SAUSA300_0386 | Xpt | 0.48 | Purines, pyrimidines, nucleosides, and nucleotides |
| SAUSA300_1258 | 0.48 | Energy metabolism | |
| SAUSA300_1293 | LysA | 0.48 | Amino acid biosynthesis |
| SAUSA300_0325 | GcvH | 0.48 | Energy metabolism |
| SAUSA300_1360 | UbiE | 0.48 | Protein synthesis |
| SAUSA300_0480 | Pth | 0.48 | Protein synthesis |
| SAUSA300_0716 | 0.48 | Purines, pyrimidines, nucleosides, and nucleotides | |
| SAUSA300_2066 | Upp | 0.47 | Purines, pyrimidines, nucleosides, and nucleotides |
| SAUSA300_0492 | FolP | 0.47 | Biosynthesis of cofactors, prosthetic groups, and carriers |
| SAUSA300_2076 | 0.46 | Central intermediary metabolism | |
| SAUSA300_1640 | Icd | 0.46 | Energy metabolism |
| SAUSA300_0841 | 0.46 | Conserved hypothetical protein | |
| SAUSA300_2197 | RplP | 0.45 | Protein synthesis |
| SAUSA300_1443 | RluB | 0.45 | Protein synthesis |
| SAUSA300_1159 | NusA | 0.45 | Transcription |
| SAUSA300_1530 | YbeY | 0.44 | Conserved hypothetical protein |
| SAUSA300_0820 | SufS | 0.44 | Biosynthesis of cofactors, prosthetic groups, and carriers |
| SAUSA300_1937 | 0.44 | Mobile and extrachromosomal element functions | |
| SAUSA300_1049 | MurI | 0.43 | Cell envelope |
| SAUSA300_1679 | AcsA | 0.43 | Central intermediary metabolism |
| SAUSA300_1178 | RecA | 0.42 | DNA metabolism |
| SAUSA300_1269 | FemA | 0.42 | Cellular processes (includes toxins and virulence factors) |
| SAUSA300_1882 | GatC | 0.41 | Signal transduction |
| SAUSA300_1614 | HemL1 | 0.41 | Biosynthesis of cofactors, prosthetic groups, and carriers |
| SAUSA300_0067 | 0.41 | Unknown function | |
| SAUSA300_1634 | CoaE | 0.40 | Biosynthesis of cofactors, prosthetic groups, and carriers |
| SAUSA300_1288 | DapA | 0.40 | Amino acid biosynthesis |
| SAUSA300_1478 | 0.37 | Cell envelope | |
| SAUSA300_1285 | 0.35 | Transport and binding proteins | |
| SAUSA300_0971 | PurL | 0.35 | Purines, pyrimidines, nucleosides, and nucleotides |
| SAUSA300_0368 | RpsR | 0.33 | Protein synthesis |
| SAUSA300_1357 | AroC | 0.33 | Amino acid biosynthesis |
| SAUSA300_0919 | MurE | 0.32 | Cell envelope |
| SAUSA300_1156 | ProS | 0.32 | Protein synthesis |
| SAUSA300_0753 | 0.30 | Conserved hypothetical protein | |
| SAUSA300_0741 | UvrB | 0.29 | DNA metabolism |
| SAUSA300_0692 | SaeQ | 0.27 | Conserved hypothetical protein |
| SAUSA300_1523 | 0.27 | Conserved hypothetical protein | |
| SAUSA300_2526 | PyrD | 0.26 | Purines, pyrimidines, nucleosides, and nucleotides |
| SAUSA300_0364 | YchF | 0.26 | Unknown function |
| SAUSA300_1144 | TrmFO | 0.24 | Unknown function |
| SAUSA300_1861 | 0.24 | Conserved hypothetical protein | |
| SAUSA300_1007 | 0.24 | Unknown function | |
| SAUSA300_0329 | 0.24 | Unknown function | |
| SAUSA300_0732 | 0.23 | Conserved hypothetical protein | |
| SAUSA300_0538 | 0.23 | Energy metabolism | |
| SAUSA300_2251 | 0.22 | Energy metabolism | |
| SAUSA300_2025 | RsbU | 0.22 | Cellular processes (includes toxins and virulence factors) |
| SAUSA300_2525 | 0.21 | Conserved hypothetical protein | |
| SAUSA300_2510 | 0.20 | Conserved hypothetical protein | |
| SAUSA300_2312 | Mqo | 0.18 | Energy metabolism |
| SAUSA300_2296 | 0.17 | Unknown function | |
| SAUSA300_0737 | SecA1 | 0.10 | Protein fate |
| SAUSA300_2477 | CidC | 0.09 | Energy metabolism |
| SAUSA300_1711 | PutA | 0.05 | Energy metabolism |
| SAUSA300_2125 | 0.05 | Transport and binding proteins | |
| SAUSA300_0857 | PpiB | 0.01 | Conserved hypothetical protein |
| Gene Designation | Protein Name | Fold Change 1 | Functional Grouping |
|---|---|---|---|
| SAUSA300_1018 | 11.13 | Conserved hypothetical protein | |
| SAUSA300_0062 | ArcB | 7.92 | Amino acid biosynthesis |
| SAUSA300_2052 | 2.96 | DNA metabolism | |
| SAUSA300_1606 | 2.63 | Conserved hypothetical protein | |
| SAUSA300_0963 | QoxA | 2.23 | Energy metabolism |
| SAUSA300_1341 | Pbp2 | 2.06 | Cell envelope |
| SAUSA300_0318 | NanE | 0.50 | Central intermediary metabolism |
| SAUSA300_1763 | EpiP | 0.44 | Protein fate |
| SAUSA300_1937 | 0.44 | Mobile and extrachromosomal element functions | |
| SAUSA300_2082 | RpoE | 0.42 | Transcription |
| SAUSA300_0923 | HtrA2 | 0.38 | Protein fate |
| SAUSA300_0279 | EsaA | 0.37 | Cell envelope |
| SAUSA300_2032 | KdpC | 0.32 | Transport and binding proteins |
| SAUSA300_0226 | FadB | 0.31 | Fatty acid and phospholipid metabolism |
| SAUSA300_1934 | 0.30 | Mobile and extrachromosomal element functions | |
| SAUSA300_1790 | PrsA | 0.03 | Protein fate |
| Gene Designation | Protein Name | Fold Change 1 | Protein Description |
|---|---|---|---|
| SAUSA300_1790 | PrsA | 15.61748634 | Foldase protein PrsA |
| SAUSA300_1512 | Pbp3 | 9.75 | Penicillin-binding protein 3 |
| SAUSA300_0838 | DltD | 7.25 | D-alanine-activating enzyme/D-alanine-D-alanyl, dltD protein |
| SAUSA300_0958 | LcpB | 6 | Transcriptional regulator |
| SAUSA300_1588 | LytH | 6 | Probable cell wall amidase LytH |
| SAUSA300_0963 | QoxA | 5.636363636 | Probable quinol oxidase subunit 2 |
| SAUSA300_1974 | LukB | 4.833333333 | Uncharacterized leukocidin-like protein 1 |
| SAUSA300_0032 | MecA | 4.5 | Penicillin-binding protein 2 |
| SAUSA300_2259 | LcpC | 4.454545455 | Putative transcriptional regulator |
| SAUSA300_0274 | 4.285714286 | Uncharacterized protein | |
| SAUSA300_0419 | 3.8125 | Uncharacterized lipoprotein | |
| SAUSA300_2136 | HtsA | 3.705882353 | Iron compound ABC transporter, iron compound-binding protein |
| SAUSA300_1982 | GroL | 3.702702703 | 60 kDa chaperonin |
| SAUSA300_0962 | QoxB | 3.608695652 | Probable quinol oxidase subunit 1 |
| SAUSA300_2578 | 3.6 | Putative phage infection protein | |
| SAUSA300_2213 | 3.5 | AcrB/AcrD/AcrF family protein | |
| SAUSA300_2328 | 3.333333333 | Uncharacterized protein | |
| SAUSA300_2092 | Dps | 3.142857143 | General stress protein 20U |
| SAUSA300_2100 | 3.133333333 | Lytic regulatory protein | |
| SAUSA300_0992 | 3.090909091 | Putative lipoprotein | |
| SAUSA300_2144 | 3.083333333 | Uncharacterized protein | |
| SAUSA300_0868 | SpsB | 3 | Signal peptidase I |
| SAUSA300_0279 | EsaA | ∞ | Putative membrane protein |
| SAUSA300_2579 | LytZ | ∞ | N-acetylmuramoyl-L-alanine amidase domain protein |
| SAUSA300 Gene Number | Protein Name | Fold Change 1 | Protein Description |
|---|---|---|---|
| SAUSA300_0857 | PpiB | 31.5 | Putative peptidyl-prolyl cis-trans isomerase |
| SAUSA300_0737 | SecA1 | 8.5 | Protein translocase subunit SecA 1 |
| SAUSA300_1027 | RpmF | 7.8 | 50S ribosomal protein L32 |
| SAUSA300_2364 | Sbi | 7.5 | Immunoglobulin-binding protein sbi |
| SAUSA300_1535 | RpsU | 6.25 | 30S ribosomal protein S21 |
| SAUSA300_1178 | RecA | 5.75 | Protein RecA |
| SAUSA300_0220 | PflB | 5.5 | Formate acetyltransferase |
| SAUSA300_1193 | GlpD | 4.88 | Aerobic glycerol-3-phosphate dehydrogenase |
| SAUSA300_1645 | PfkA | 4.83 | ATP-dependent 6-phosphofructokinase |
| SAUSA300_0757 | Pgk | 4.83 | Phosphoglycerate kinase |
| SAUSA300_0798 | 4.63 | Lipoprotein | |
| SAUSA300_0388 | GuaB | 4.5 | Inosine-5′-monophosphate dehydrogenase |
| SAUSA300_1525 | GlyQS | 4.18 | Glycine--tRNA ligase |
| SAUSA300_0489 | FtsH | 3.86 | ATP-dependent zinc metalloprotease FtsH |
| SAUSA300_2067 | GlyA | 3.75 | Serine hydroxymethyltransferase |
| SAUSA300_1880 | GatB | 3.63 | Aspartyl/glutamyl-tRNA (Asn/Gln) amidotransferase subunit B |
| SAUSA300_0491 | CysK | 3.63 | Cysteine synthase |
| SAUSA300_1150 | Tsf | 3.62 | Elongation factor Ts |
| SAUSA300_0389 | GuaA | 3.56 | GMP synthase [glutamine-hydrolyzing] |
| SAUSA300_1684 | 3.25 | Uncharacterized protein | |
| SAUSA300_0533 | Tuf | 3.09 | Elongation factor Tu |
| SAUSA300_0693 | SaeP | 3.07 | Putative lipoprotein |
| SAUSA300_1459 | Gnd | 3.06 | 6-phosphogluconate dehydrogenase, decarboxylating |
| SAUSA300_0496 | LysS | 3 | Lysine--tRNA ligase |
| SAUSA300_0716 | NrdE | ∞ | Ribonucleoside-diphosphate reductase |
| SAUSA300_2251 | ∞ | Dehydrogenase family protein | |
| SAUSA300_1881 | GatA | ∞ | Glutamyl-tRNA (Gln) amidotransferase subunit A |
| SAUSA300_1586 | AspS | ∞ | Aspartate--tRNA ligase |
| SAUSA300_1167 | Pnp | ∞ | Polyribonucleotide nucleotidyltransferase |
| SAUSA300_0009 | SerS | ∞ | Serine--tRNA ligase |
| SAUSA300_1629 | ThrS | ∞ | Threonine--tRNA ligase |
| SAUSA300_2214 | FemX | ∞ | Lipid II:glycine glycyltransferase |
| Name | Characteristics | Source |
|---|---|---|
| Strains | ||
| S. aureus | ||
| RN4220 | Restriction-deficient transformation recipient | [43] |
| TCH1516 | Community-associated USA300 MRSA isolate | [44] |
| RKC0323 | TCH1516 ΔppiB | [21] |
| RKC0085 | TCH1516 ΔprsA | [24] |
| RKC0183 | TCH1516 hla::Bursa, hla mutant | (21) |
| RKC0374 | TCH1516 ΔppiB pMK4_ppiB-HA | This work |
| RKC0536 | TCH1516 ΔppiB pMK4 | This work |
| RKC0283 | TCH1516 ΔprsA pMK4_prsA-his | (24) |
| RKC0129 | TCH1516 ΔprsA pMK4 | (24) |
| JE2 | USA300 LAC isolate cured of plasmids LAC-p01 and LAC-p03 | [45] |
| NE1354 | USA300 JE2 hla::Bursa, hla NTML transposon mutant | [45] |
| AH1263 | USA300 Lac isolate cured of plasmid Lac-p03 | [46] |
| RKC0521 | AH1263 hla::Bursa, hla mutant | [47] |
| RKC0753 | AH1263 ΔαPSMs | This work |
| Plasmids | ||
| pMK4 | Gram-positive shuttle vector (Cmr) | [48] |
| pRKC0131 | pMK4_ppiB-HA (vector overexpressing ppiB with an HA tag) | [24] |
| pRKC0126 | pMK4_prsA-His (vector overexpressing prsA with a poly-histidine tag) | [24] |
| pRKC0674 | pJB38 containing DNA flanking αPSM transcript with ery cassette | This work |
| Name | Sequence |
|---|---|
| #0273 | GGTGCTGGGCAAATACAAGT (gyrB) |
| #0274 | TCCCACACTAAATGGTGCAA (gyrB) |
| #0263 | TGCAAATGTTTCGATTGGTC (hla) |
| #0264 | CCCCAATTTTGATTCACCATA (hla) |
| #0271 | ACAGGAGGACAAAACGATGG (psmα) |
| #0272 | CCCTATTGGTATAGTGGCCTGA (psmα) |
| #0490 | CAAGACGTCCGTCGGTCTACCTTTCCATGC |
| #0493 | GGGGTACCACGTGGCACTTTCCAAAAAC |
| #0638 | CCGGAATTCGCTCCTTGGAAGCTGTCAGT |
| #0639 | AAAACTGCAGGAAGCAAACTTAAGAGTGTGTTGA |
| #0644 | CCGGAATTCGATGTGAGGTGAGTCTTGTTAGTTTG |
| #0645 | AAAACTGCAGAGATTACCTCCTTTGCTTATGAGT |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keogh, R.A.; Zapf, R.L.; Trzeciak, E.; Null, G.G.; Wiemels, R.E.; Carroll, R.K. Novel Regulation of Alpha-Toxin and the Phenol-Soluble Modulins by Peptidyl-Prolyl cis/trans Isomerase Enzymes in Staphylococcus aureus. Toxins 2019, 11, 343. https://doi.org/10.3390/toxins11060343
Keogh RA, Zapf RL, Trzeciak E, Null GG, Wiemels RE, Carroll RK. Novel Regulation of Alpha-Toxin and the Phenol-Soluble Modulins by Peptidyl-Prolyl cis/trans Isomerase Enzymes in Staphylococcus aureus. Toxins. 2019; 11(6):343. https://doi.org/10.3390/toxins11060343
Chicago/Turabian StyleKeogh, Rebecca A., Rachel L. Zapf, Emily Trzeciak, Gillian G. Null, Richard E. Wiemels, and Ronan K. Carroll. 2019. "Novel Regulation of Alpha-Toxin and the Phenol-Soluble Modulins by Peptidyl-Prolyl cis/trans Isomerase Enzymes in Staphylococcus aureus" Toxins 11, no. 6: 343. https://doi.org/10.3390/toxins11060343
APA StyleKeogh, R. A., Zapf, R. L., Trzeciak, E., Null, G. G., Wiemels, R. E., & Carroll, R. K. (2019). Novel Regulation of Alpha-Toxin and the Phenol-Soluble Modulins by Peptidyl-Prolyl cis/trans Isomerase Enzymes in Staphylococcus aureus. Toxins, 11(6), 343. https://doi.org/10.3390/toxins11060343