The In Vivo and In Vitro Toxicokinetics of Citreoviridin Extracted from Penicillium citreonigrum

Abstract
:1. Introduction
2. Results
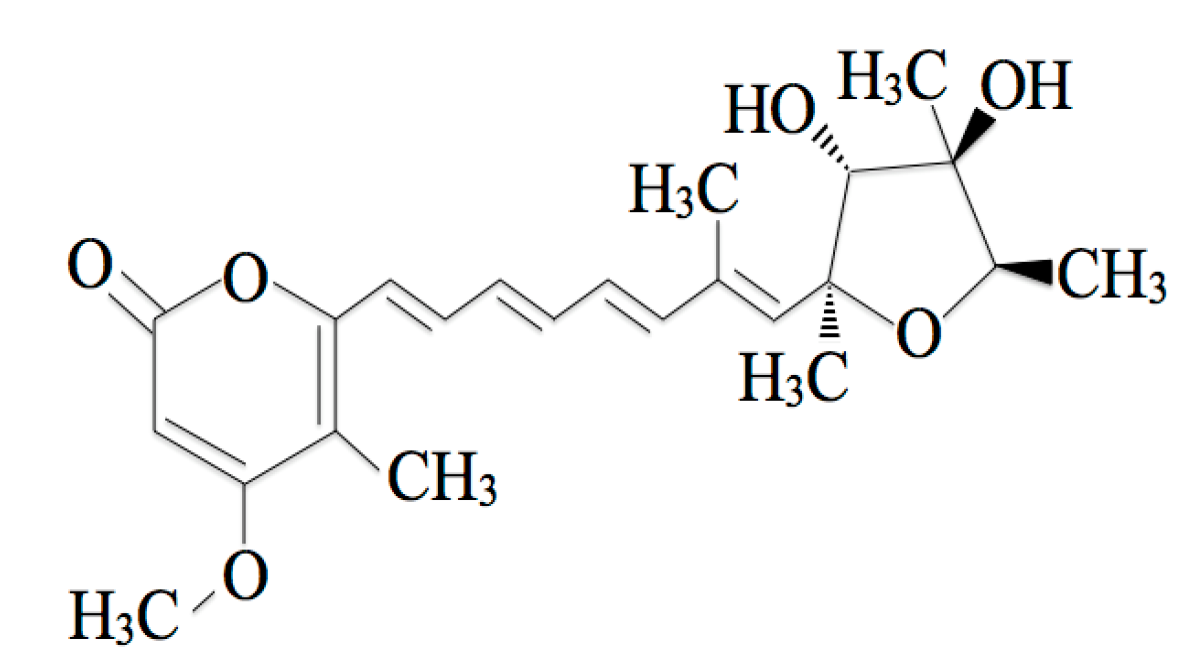
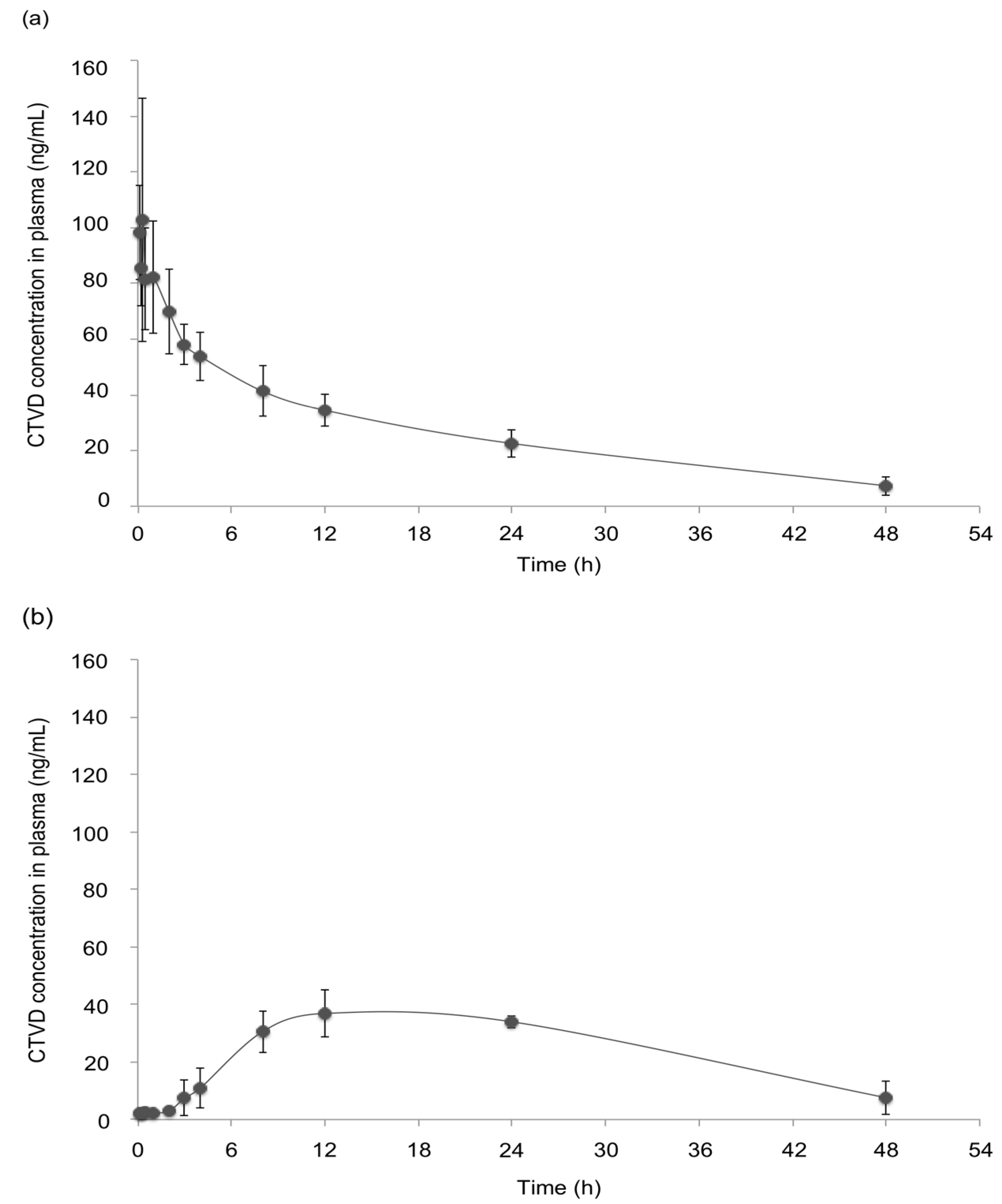
2.1. The Toxicokinetics of CTVD in Swine
2.2. Permeability Study Using Caco-2 Cells
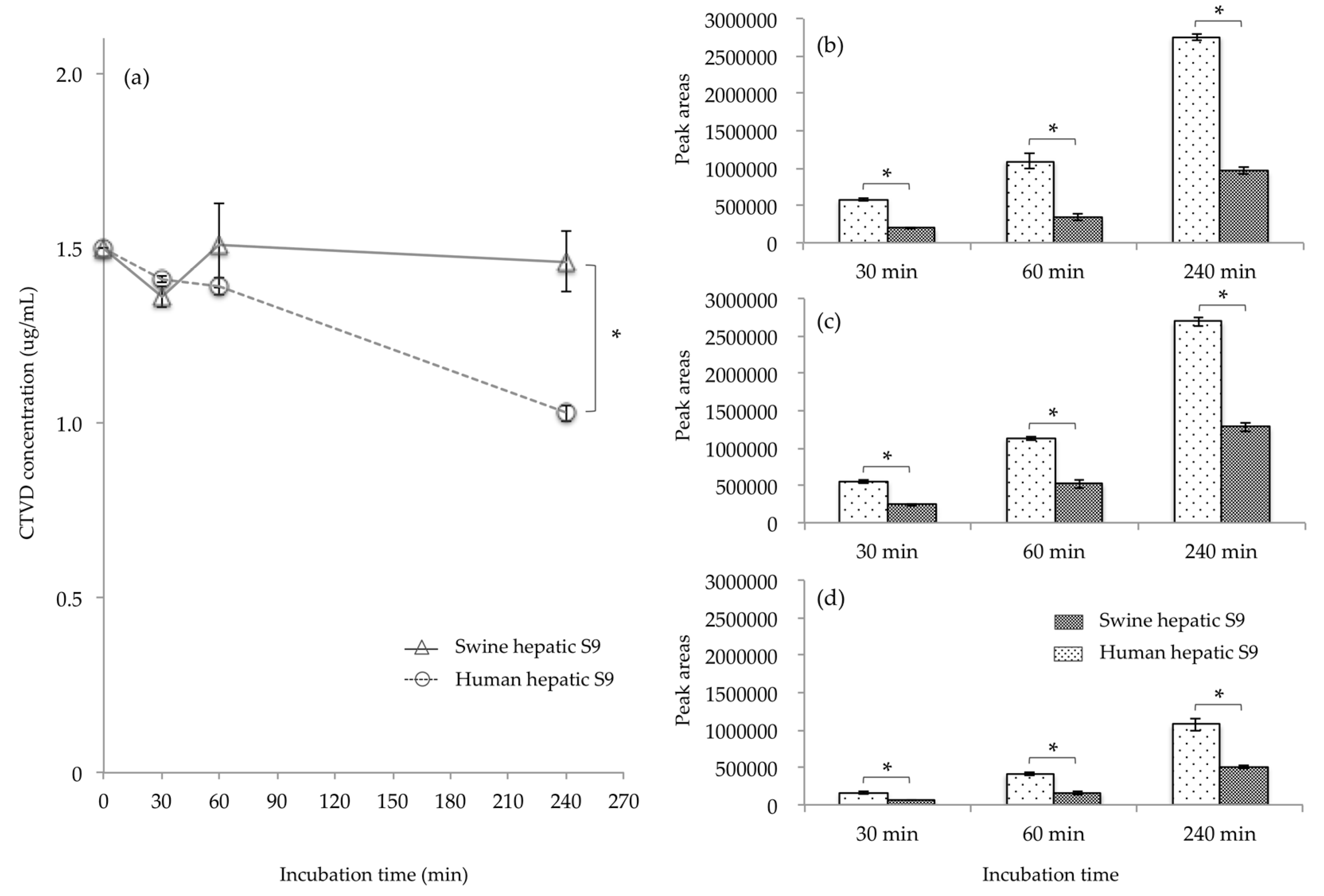
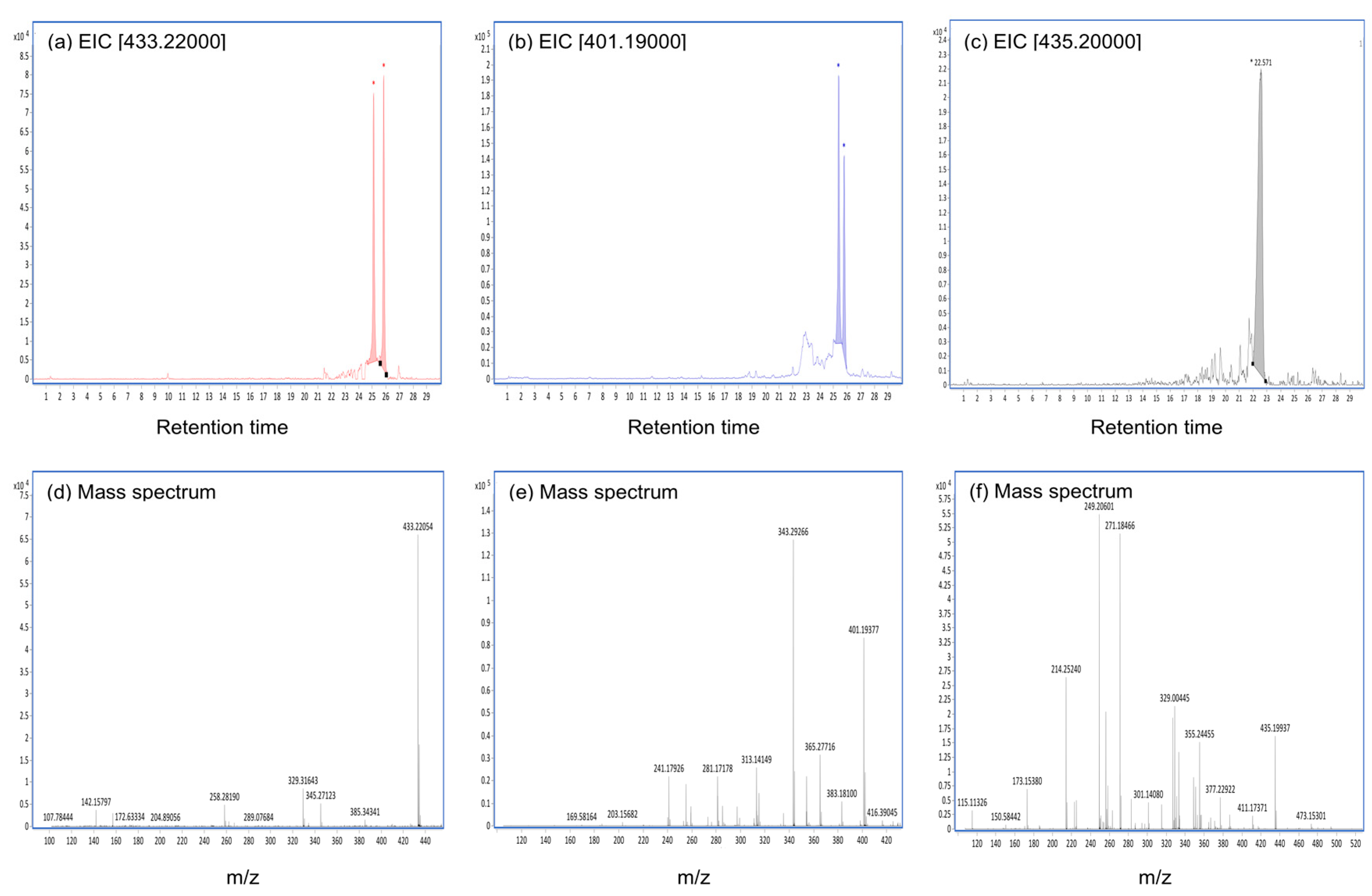
2.3. CTVD Elimination and the CTVD Metabolite Profile Following Incubation with S9 Fraction In Vitro
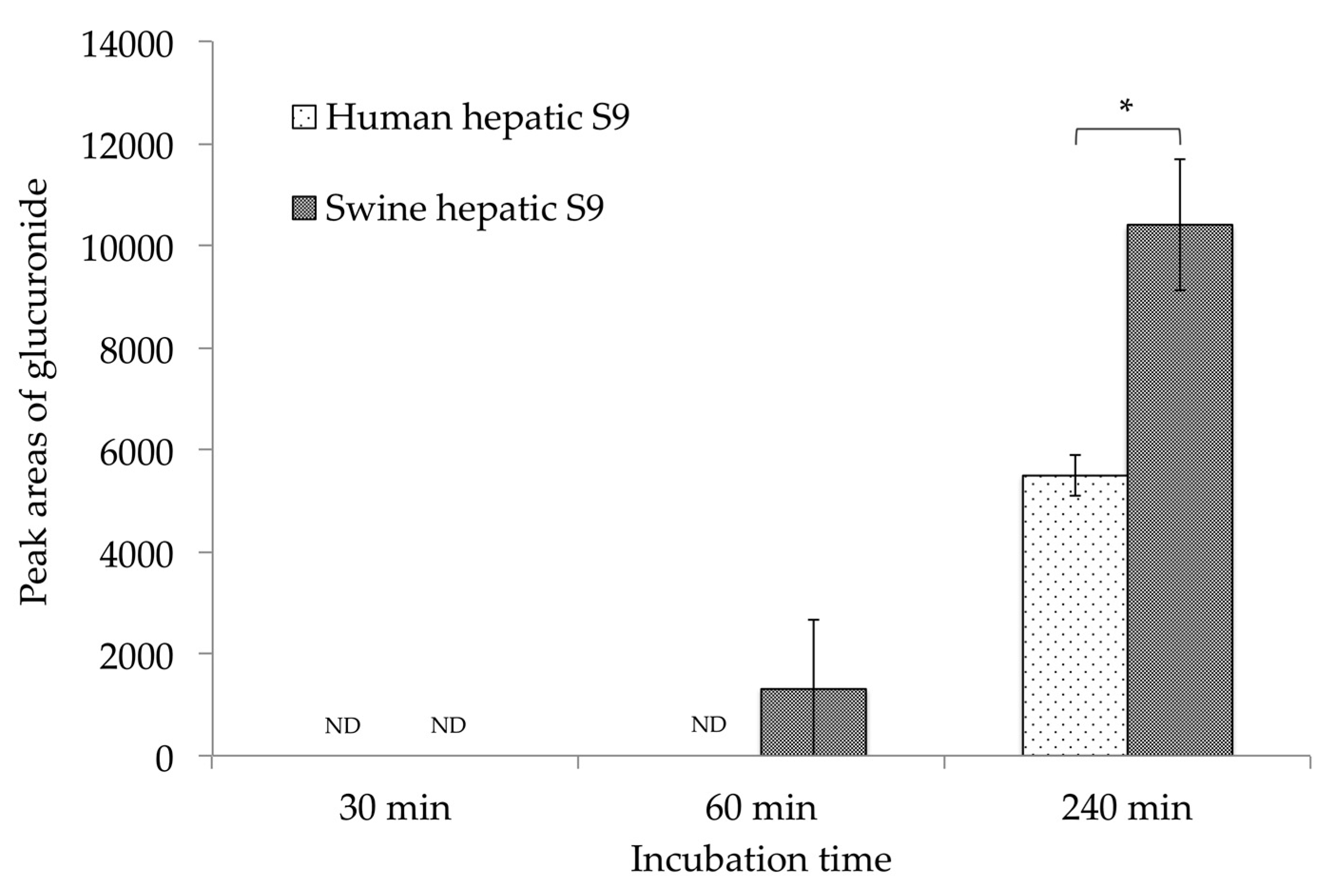
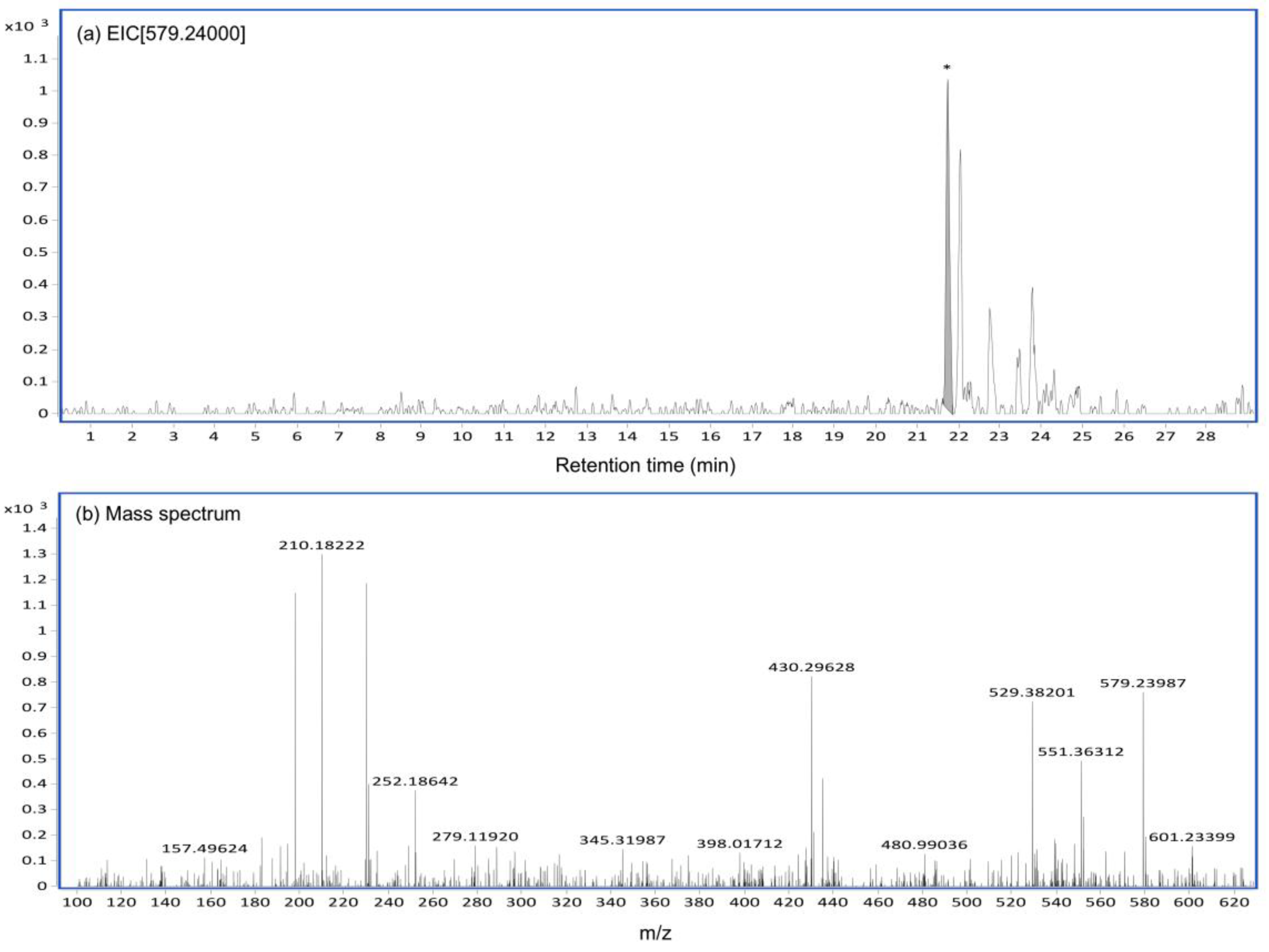
2.4. CTVD Elimination and the CTVD Glucuronide Profile Following Incubation with S9 In Vitro
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Administration Study
4.2.1. Animals and Diets
4.2.2. Administration and Blood Sampling
4.2.3. Toxicokinetic Analyses
4.3. Permeability Study Using Caco-2 Cells
4.4. Production of CTVD Metabolites by Incubating with S9 Fractions
4.5. Quantification of CTVD and Detection of Metabolites
4.6. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolites | Retention Time (min) | Polarity | Base Peak Ion | Mass Accuracy |
|---|---|---|---|---|
| Relative Mass Error | ||||
| Hydroxylation and methylation | 25.10 | Positive | (M + H)+ | 2.9 |
| Desaturation | 25.79 | Positive | (M + H)+ | 1.1 |
| Dihydroxylation | 22.57 | Positive | (M + H)+ | 3.1 |
| Name | Chemical Formula | Exact Mass | Name | Chemical Formula | Exact Mass |
|---|---|---|---|---|---|
| (O, N, S) Methylation | C24H32O6 | 416.21989 | Ethyl to Carboxylic Acid | C22H26O8 | 418.16277 |
| 1,4-Dihydropyridines to Pyridines | C23H28O6 | 400.18859 | First/Second Alcohols to Aldehyde/Ketone | C23H28O6 | 400.18859 |
| 2-Ethoxyl to Acid | C22H26O7 | 402.16785 | Glucuronidation + Hydrogenation | C29H40O12 | 580.25198 |
| 2 × Glucuronide Conjugation | C35H46O18 | 754.26841 | Glucuronide Conjugation | C29H38O12 | 578.23633 |
| 2 × Hydrogenation | C23H34O6 | 406.23554 | Glutamine Conjugation | C28H38NO8 | 516.25974 |
| 2 × Hydroxylation and Sulfation | C23H30O14S2 | 594.10770 | Gluthation Conjugation | C33H47N3O12S | 709.28804 |
| 2 × Hydroxylation | C23H30O8 | 434.19407 | Gluthation Conjugation + Demethylation | C32H43N3O12S | 693.25674 |
| 2 × Hydroxylation + 2 × Glucuronide | C35H46O20 | 786.25824 | Gluthation Conjugation + Dihydroxylation | C33H45N3O14S | 739.26222 |
| 2 × Oxidation + Glucuronidation | C29H38O14 | 610.22616 | Gluthation Conjugation + Hydroxylation | C33H45N3O13S | 723.26731 |
| 2 × Sulfate Conjugation | C23H30O12S2 | 562.11787 | Gluthation Conjugation, Hydroxylation + Oxidation | C33H43N3O14S | 737.24657 |
| 3 × Hydroxylation | C23H30O9 | 450.18898 | Glycine Conjugation | C25H33NO7 | 459.22570 |
| 3 × Oxidation + Dehydrogenation | C23H28O9 | 448.17333 | Hetero oxide reduction + Hydrogenation | C23H32O5 | 388.22497 |
| Acetylation | C25H32O7 | 444.21480 | Hydration, Hydrolysis (Internal) | C23H32O7 | 420.21480 |
| Acetylation + Oxidation | C25H32O8 | 460.20972 | Hydrogenation | C23H32O6 | 404.21989 |
| Alcohols Dehydration | C23H28O5 | 384.19367 | Hydrolysis + 2× Oxidation | C23H32O9 | 452.20463 |
| Alkene to Epoxide | C23H30O7 | 418.19915 | Hydroxylation | C23H30O7 | 418.19915 |
| Alkenes to Dihydrodiol | C23H32O8 | 436.20972 | Hydroxylation + Glucuronide | C29H38O13 | 594.23124 |
| Aromatic Oxidation | C24H32O8 | 448.20972 | Hydroxylation and Dehydration | C23H28O6 | 400.18859 |
| Aromatic Ring to Arene Oxide | C23H30O7 | 418.19915 | Hydroxylation and Desaturation | C23H28O7 | 416.18350 |
| Carboxylation + Glucuronidation | C29H36O14 | 608.21051 | Hydroxylation and Ketone Formation | C23H28O8 | 432.17842 |
| Cysteine Conjugation | C26H37NO8S | 523.22399 | Hydroxylation and Methylation | C24H32O7 | 432.21480 |
| Cysteine Conjugation and Desaturation | C26H35NO8S | 521.20834 | Hydroxylation and Sulfation | C23H30O10S | 498.15597 |
| Cysteine Glycine Conjugation | C28H40N2O9S | 580.24545 | Hydroxymethylene Loss | C22H28O5 | 372.19367 |
| Cysteine Glycine Conjugation and Desaturation | C28H38N2O9S | 578.22980 | Isopropyl Dealkylation | C20H24O6 | 360.15729 |
| Deacetylation | C21H28O5 | 360.19367 | Isopropyl to Acid | C21H24O8 | 404.14712 |
| Dacetylation + Dehydrogenation | C21H26O5 | 358.17802 | Isopropyl to Alcohol | C20H24O7 | 376.15220 |
| Debenzylation | C16H24O6 | 312.15729 | Ketone to Alcohol | C23H32O6 | 404.21989 |
| Debutylation + Hydrogenation | C19H24O6 | 348.15729 | Methyl Ketone to Acid | C21H26O7 | 390.16785 |
| Decarbonylation | C22H30O5 | 374.20932 | Methylene to Ketone | C23H28O7 | 416.18350 |
| Decarboxylation | C22H30O4 | 358.21441 | N-Acethylcysteine Conjugation | C28H39NO9S | 565.23455 |
| Decarboxylation and Glucuronidation | C28H38O11 | 550.24141 | N-Acethylcysteine Conjugation and Desaturation | C28H37NO9S | 563.21890 |
| Deethylation | C21H26O6 | 374.17294 | Oxidation + 2× Desaturation | C23H26O7 | 414.16785 |
| Demethylation | C22H28O6 | 388.18859 | Oxidation + Acethylcysteination | C28H39NO10S | 581.22947 |
| Demethylation + Dehydrogenation | C22H26O6 | 386.17294 | Oxidation + Deacetylation | C21H28O6 | 376.18859 |
| Demethylation + Glucuronidation | C28H36O12 | 564.22068 | Oxidation + Demethylation + Dehydrogenation | C22H26O7 | 402.16785 |
| Demethylation + Hydrogenation | C22H30O6 | 390.20424 | Parent | C23H30O6 | 402.20424 |
| Demethylation + Oxidation + Glucuronidation | C28H36O13 | 580.21559 | Phosphorylation | C23H31O9P | 482.17057 |
| Demethylation and Hydroxylation | C22H28O7 | 404.18350 | Propyl Ether to Acid | C20H22O7 | 374.13655 |
| Demethylation and Methylene to Ketone | C22H26O7 | 402.16785 | Propyl Ketone to Acid | C19H22O7 | 362.13655 |
| Demethylation and two Hydroxylations | C22H28O8 | 420.17842 | Quinone Formation | C23H28O8 | 432.17842 |
| Demethylation to Carboxylic Acid | C23H28O8 | 432.17842 | Sulfate Conjugation | C23H30O9S | 482.16105 |
| Desaturation | C23H28O6 | 400.18859 | Taurine Conjugation | C25H35NO8S | 509.20834 |
| Desaturation + Gluthation Conjugation | C33H45N3O12S | 707.27239 | Tert-Butyl Dealkylation | C19H22O6 | 346.14164 |
| Epoxidation + Gluthation Conjugation | C33H47N3O13S | 725.28296 | Tert-Butyl to Acid | C20H22O8 | 390.13147 |
| Ethyl Ether to Acid | C21H24O7 | 388.15220 | Tert-Butyl to Alcohol | C19H22O7 | 362.13655 |
| Ethyl Ketone to Acid | C20H24O7 | 376.15220 | Triphosphorylation | C23H33O15P3 | 642.10323 |
| Ethyl to Alcohol | C21H26O7 | 390.16785 | Two Sequential Desaturations | C23H26O6 | 398.17294 |


References
- Lima, H.C.A.V.; Porto, E.A.S.; Marins, J.R.P.; Alves, R.M.; Machado, R.R.; Braga, K.N.L.; de Paiva, B.; Carmo, G.M.I.; Faria Silva e Santelli, A.C.; Sobel, J. Outbreak of beriberi in the state of Maranhão, Brazil: Revisiting the mycotoxin aetiologic hypothesis. Trop. Dr. 2010, 40, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Cole, R.J.; Dorner, J.W.; Cox, R.H.; Hill, R.A.; Cluter, H.G.; Wells, J.M. Isolation of citreoviridin from Penicillium charlesii cultures and molded pecan fragments. Appl. Environ. Microbiol. 1981, 42, 677–681. [Google Scholar] [PubMed]
- Wicklow, D.T.; Cole, R.J. Citreoviridin in standing corn infested by Eupenicillium ochrosalmoneum. Mycologia 1984, 76, 959–961. [Google Scholar] [CrossRef]
- Wicklow, D.T.; Stubblefield, R.D.; Horn, B.W.; Shotwell, O.L. Citreoviridin levels in Eupenicillium ochrosalmoneum-infested maize kernels at harvest. Appl. Environ. Microbiol. 1988, 54, 1096–1098. [Google Scholar] [PubMed]
- Almeida, M.I.; Almeida, N.G.; Carvalho, K.L.; Gonçalves, G.A.A.; Silva, C.N.; Santos, E.A.; Garcia, J.C.; Vargas, E.A. Co-occurrence of aflatoxins B1, B2, G1 and G2, ochratoxin A, zearalenone, deoxynivalenol, and citreoviridin in rice in Brazil. Food Addit. Contam. Part A 2012, 29, 694–703. [Google Scholar] [CrossRef]
- Shiratori, N.; Kobayashi, N.; Tulayakul, P.; Sugiura, Y.; Takino, M.; Endo, O.; Sugita-Konishi, Y. Occurrence of Penicillium brocae and Penicillium citreonigrum, which produce a mutagenic metabolite and a mycotoxin citreoviridin, respectively, in selected commercially available rice grains in Thailand. Toxins 2017, 9, 194. [Google Scholar] [CrossRef]
- Kushiro, M. Historical review of researches on yellow rice and mycotoxigenic fungi adherent to rice in Japan. JSM Mycotoxins 2015, 65, 19–23. [Google Scholar] [CrossRef] [Green Version]
- Ueno, Y. Temperature-dependent production of citreoviridin, a neurotoxin of Penicillium citreo-viride Biourge. Jpn. J. Exp. Med. 1972, 42, 107–114. [Google Scholar]
- Padilha, E.M.; Fujimori, E.; Borges, A.L.; Sato, A.P.; Gomes, M.N.; Branco, M.R.; Santos, H.J.; Lermen, N., Jr. Epidemiological profile of reported beriberi cases in Maranhão State, Brazil, 2006–2008. Cad. Saude Publica 2011, 27, 449–459. [Google Scholar] [CrossRef]
- Rosa, C.A.R.; Keller, K.M.; Oliveira, A.A.; Almeida, T.X.; Keller, L.A.M.; Marassi, A.C.; Kruger, C.D.; Deveza, M.V.; Monteiro, B.S.; Nunes, L.M.T.; et al. Production of citreoviridin by Penicillium citreonigrum strains associated with rice consumption and beriberi cases in the Maranhão State, Brazil. Food Addit. Contam. A 2010, 27, 241–248. [Google Scholar] [CrossRef]
- Uraguchi, K. Mycotoxic origin of cardiac beriberi. J. Stored Prod. Res. 1969, 5, 227–236. [Google Scholar] [CrossRef]
- Ueno, Y.; Ueno, I. Isolation and acute toxicity of citreoviridin, a neurotoxic mycotoxin of Penicillium citreo-viride Biourge. Jpn. J. Exp. Med. 1972, 42, 91–105. [Google Scholar] [PubMed]
- Ueno, Y. Citreoviridin from Penicillium citreoviride Biourge. In Mycotoxins; Purchase, I.F.H., Ed.; Elsevier Scientific Publ. Co.: New York, NY, USA, 1974; pp. 283–302. [Google Scholar]
- Nishie, K.; Cole, R.J.; Dorner, J.W. Toxicity of citreoviridin. Res. Commun. Chem. Pathol. Pharmacol. 1988, 59, 31–52. [Google Scholar] [PubMed]
- Uraguchi, K. Evidence of a toxin in the yellowed rice polluted by Penicillium sp. Pharmacological studies on the toxicity of the yellowed rice “O-hen mai” I. Nisshin. Igaku 1947, 34, 155–161. [Google Scholar]
- Sakai, F.; Uraguchi, K. Studies by long-term feeding experiments with rats on development of chronic poisoning by toxic substance from yellowsis rice. VII. Pharmacological studies on toxicity of yellowsis rice. Nisshin. Igaku 1955, 42, 609–617. [Google Scholar]
- Sun, S. Chronic exposure to cereal mycotoxin likely citreoviridin may be a trigger for Keshan disease mainly through oxidative stress mechanism. Med. Hypotheses 2010, 74, 841–842. [Google Scholar] [CrossRef]
- Lau, Y.Y.; Chen, Y.H.; Liu, T.T.; Li, C.; Cui, X.; White, R.E.; Cheng, K.C. Evaluation of a novel in vitro Caco-2 hepatocyte hybrid system for predicting in vivo oral bioavailability. Drug Metab. Dispos. 2004, 32, 937–942. [Google Scholar]
- Li, C.; Liu, T.; Cui, X.; Uss, A.S.; Cheng, K.C. Development of in vitro pharmacokinetic screens using Caco-2, human hepatocyte, and Caco-2/human hepatocyte hybrid systems for the prediction of oral bioavailability in humans. J. Biomol. Screen. 2007, 12, 1084–1091. [Google Scholar]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER measurement techniques for in vitro barrier model systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef]
- Kadota, T.; Furusawa, H.; Hirano, S.; Tajima, O.; Kamata, Y.; Sugita-Konishi, Y. Comparative study of deoxynivalenol, 3-acetyldeoxynivalenol, and 15-acetyldeoxynivalenol on intestinal transport and IL-8 secretion in the human cell line Caco-2. Toxicol. In Vitro 2013, 27, 1888–1895. [Google Scholar] [CrossRef]
- Jaeg, J.P.; Perdu, E.; Dolo, L.; Debrauwer, L.; Cravedi, J.P.; Zalko, D. Characterization of new bisphenol A metabolites produced by CD1 mice liver microsomes and S9 fractions. J. Agric. Food Chem. 2004, 52, 4935–4942. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, B.; Løkken, T.; Zahlsen, K.; Nilsen, O.G. Comparison and in vivo relevance of two different in vitro head space metabolic systems: Liver S9 and liver slices. Pharmacol. Toxicol. 1997, 81, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.N.; McKown, L.A. In Vitro Drug Metabolite Profiling Using Hepatic S9 and Human Liver Microsomes. In Optimization in Drug Discovery; Yan, Z., Caldwell, G.W., Eds.; Humana Press: New York, NY, USA, 2004; pp. 163–184. [Google Scholar]
- Merrifield, C.A.; Lewis, M.; Claus, S.P.; Beckonert, O.P.; Dumas, M.E.; Duncker, S.; Kochhar, S.; Rezzi, S.; Lindon, J.C.; Bailey, M.; et al. A metabolic system-wide characterisation of the pig: A model for human physiology. Mol. Biosyst. 2011, 7, 2577–2588. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Oyama, M.; Nishikawa, M.; Ikushiro, S.; Hara, H. Simultaneous collection of the portal and superior vena cava blood in conscious rats defined that intestinal epithelium is the major site of glucuronidation, but not sulfation and methylation, of quercetin. Biosci. Biotechnol. Biochem. 2018, 82, 2118–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyarts, T.; Dänicke, S. Bioavailability of the Fusarium toxin deoxynivalenol (DON) from naturally contaminated wheat for the pig. Toxicol. Lett. 2006, 163, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Prelusky, D.B.; Hartin, K.E.; Trenholm, H.L.; Miller, J.D. Pharmacokinetic fate of 14C-labeled deoxynivalenol in swine. Toxicol. Sci. 1988, 10, 276–286. [Google Scholar] [CrossRef]
- Rohweder, D.; Kersten, S.; Valenta, H.; Sondermann, S.; Schollenberger, M.; Drochner, W.; Dänicke, S. Bioavailability of the Fusarium toxin deoxynivalenol (DON) from wheat straw and chaff in pigs. Arch. Anim. Nutr. 2013, 67, 37–47. [Google Scholar] [CrossRef]
- Galtier, P.; Alvinerie, M.; Charpenteau, J.L. The pharmacokinetic profiles of ochratoxin A in pigs, rabbits and chickens. Food Cosmet. Toxicol. 1981, 19, 735–738. [Google Scholar] [CrossRef]
- Biehl, M.L.; Prelusky, D.B.; Koritz, G.D.; Hartin, K.E.; Buck, W.B.; Trenholm, H.L. Biliary excretion and enterohepatic cycling of zearalenone in immature pigs. Toxicol. Appl. Pharmacol. 1993, 121, 152–159. [Google Scholar] [CrossRef]
- Yiannikouris, A.; Jouany, J.P. Mycotoxins in feeds and their fate in animals: A review. Anim. Res. 2002, 51, 81–99. [Google Scholar] [CrossRef]
- Charman, W.N.; Stella, V.J. Transport of lipophilic molecules by the intestinal lymphatic system. Adv. Drug Deliv. Rev. 1991, 7, 1–14. [Google Scholar] [CrossRef]
- Artursson, P.E.R. Epithelial transport of drugs in cell culture. I: A model for studying the passive diffusion of drugs over intestinal absorbtive (Caco-2) cells. J. Pharm. Sci. 1990, 79, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Van De Walle, J.; Sergent, T.; Piront, N.; Toussaint, O.; Schneider, Y.J.; Larondelle, Y. Deoxynivalenol affects in vitro intestinal epithelial cell barrier integrity through inhibition of protein synthesis. Toxicol. Appl. Pharmacol. 2010, 245, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, E.; Kommer, A.; Dempe, J.S.; Hildebrand, A.A.; Metzler, M. Absorption and metabolism of the mycotoxin zearalenone and the growth promotor zeranol in Caco-2 cells in vitro. Mol. Nutr. Food Res. 2011, 55, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Caloni, F.; Cortinovis, C.; Pizzo, F.; De Angelis, I. Transport of aflatoxin M1 in human intestinal Caco-2/TC7 cells. Front. Pharmacol. 2012, 3, 111. [Google Scholar] [CrossRef] [PubMed]
- Leibholz, J. Digestion in the pig between 7 and 35 d of age 6. The digestion of hydrolyzed milk and soya-bean proteins. Br. J. Nutr. 1981, 46, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rychlik, M.; Kircher, F.; Schusdziarra, V.; Lippl, F. Absorption of the mycotoxin patulin from the rat stomach. Food Chem. Toxicol. 2004, 42, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Dänicke, S.; Valenta, H.; Döll, S. On the toxicokinetics and the metabolism of deoxynivalenol (DON) in the pig. Arch. Anim. Nutr. 2004, 58, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Baggot, J.D. Principles of Drug Disposition in Domestic Animals: The Basis of Veterinary Clinical Pharmacology; W.B. Saunders Co.: Philadelphia, PA, USA, 1977; pp. 114–145. [Google Scholar]
- Hagelberg, S.; Hult, K.; Fuchs, R. Toxicokinetics of ochratoxin A in several species and its plasma-binding properties. J. Appl. Toxicol. 1989, 9, 91–96. [Google Scholar] [CrossRef]
- Coulombe, R.A., Jr.; Sharma, R.P. Clearance and excretion of intratracheally and orally administered aflatoxin B1 in the rat. Food Chem. Toxicol. 1985, 23, 827–830. [Google Scholar] [CrossRef]
- Prelusky, D.B.; Trenholm, H.L.; Savard, M.E. Pharmacokinetic fate of 14C-labelled fumonisin B1 in swine. Nat. Toxins 1994, 2, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Qu, X.; Li, Y.; Kong, Y.; Jia, B.; Yao, X.; Jiang, B. Binding of citreoviridin to human serum albumin: Multispectroscopic and molecular docking. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Stojković, R.; Hult, K.; Gamulin, S.; Plestina, R. High affinity binding of ochratoxin A to plasma constituents. Biochem. Int. 1984, 9, 33–38. [Google Scholar] [PubMed]
- Kőszegi, T.; Poór, M. Ochratoxin A: Molecular interactions, mechanisms of toxicity and prevention at the molecular level. Toxins 2016, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Maul, R.; Warth, B.; Kant, J.S.; Schebb, N.H.; Krska, R.; Koch, M.; Sulyok, M. Investigation of the hepatic glucuronidation pattern of the Fusarium mycotoxin deoxynivalenol in various species. Chem. Res. Toxicol. 2012, 25, 2715–2717. [Google Scholar] [CrossRef] [PubMed]
- da Rocha, M.W.; Resck, I.S.; Caldas, E.D. Purification and full characterisation of citreoviridin produced by Penicillium citreonigrum in yeast extract sucrose (YES) medium. Food Addit. Contam. A 2015, 32, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Devreese, M.; De Baere, S.; De Backer, P.; Croubels, S. Quantitative determination of several toxicological important mycotoxins in pig plasma using multi-mycotoxin and analyte-specific high performance liquid chromatography–tandem mass spectrometric methods. J. Chromatogr. A 2012, 1257, 74–80. [Google Scholar] [CrossRef]




| BW (kg) | Kel ×10−1 (h−1) | T1/2 (h) | Vd or Vd/F (L) | MRT (h) | AUCt (h·ng/mL) | AUC∞ (h·ng/mL) | Cmax (ng/mL) | Tmax (h) | Ft (%) | F (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| IV | 9.4 ± 1.3 | 0.5 ± 0.1 | 16.2 ± 4.3 | 1.5 ± 0.2 | 14.6 ± 1.2 | 1322.2 ± 224.4 | 1512.9 ± 331.7 | - | - | - | - |
| PO | 10.7 ± 1.3 | 0.4 ± 0.2 | 21.4 ± 12.7 | 1.7 ± 0.3 | 19.6 ± 4.0 | 1048.2 ± 180.8 | 1761.1 ± 813.5 | 38.2 ± 6.7 | 15.0 ± 6.0 | 79.3 | 116.4 |
| Parameter | 3 µmol/L | 10 µmol/L |
|---|---|---|
| Papp (×10−6 cm/s) | 52.2 ± 28.3 | 42.6 ± 17.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uchiyama, Y.; Takino, M.; Noguchi, M.; Shiratori, N.; Kobayashi, N.; Sugita-Konishi, Y. The In Vivo and In Vitro Toxicokinetics of Citreoviridin Extracted from Penicillium citreonigrum. Toxins 2019, 11, 360. https://doi.org/10.3390/toxins11060360
Uchiyama Y, Takino M, Noguchi M, Shiratori N, Kobayashi N, Sugita-Konishi Y. The In Vivo and In Vitro Toxicokinetics of Citreoviridin Extracted from Penicillium citreonigrum. Toxins. 2019; 11(6):360. https://doi.org/10.3390/toxins11060360
Chicago/Turabian StyleUchiyama, Yosuke, Masahiko Takino, Michiko Noguchi, Nozomi Shiratori, Naoki Kobayashi, and Yoshiko Sugita-Konishi. 2019. "The In Vivo and In Vitro Toxicokinetics of Citreoviridin Extracted from Penicillium citreonigrum" Toxins 11, no. 6: 360. https://doi.org/10.3390/toxins11060360
APA StyleUchiyama, Y., Takino, M., Noguchi, M., Shiratori, N., Kobayashi, N., & Sugita-Konishi, Y. (2019). The In Vivo and In Vitro Toxicokinetics of Citreoviridin Extracted from Penicillium citreonigrum. Toxins, 11(6), 360. https://doi.org/10.3390/toxins11060360