Cytotoxicity Produced by Silicate Nanoplatelets: Study of Cell Death Mechanisms

 , and
, and 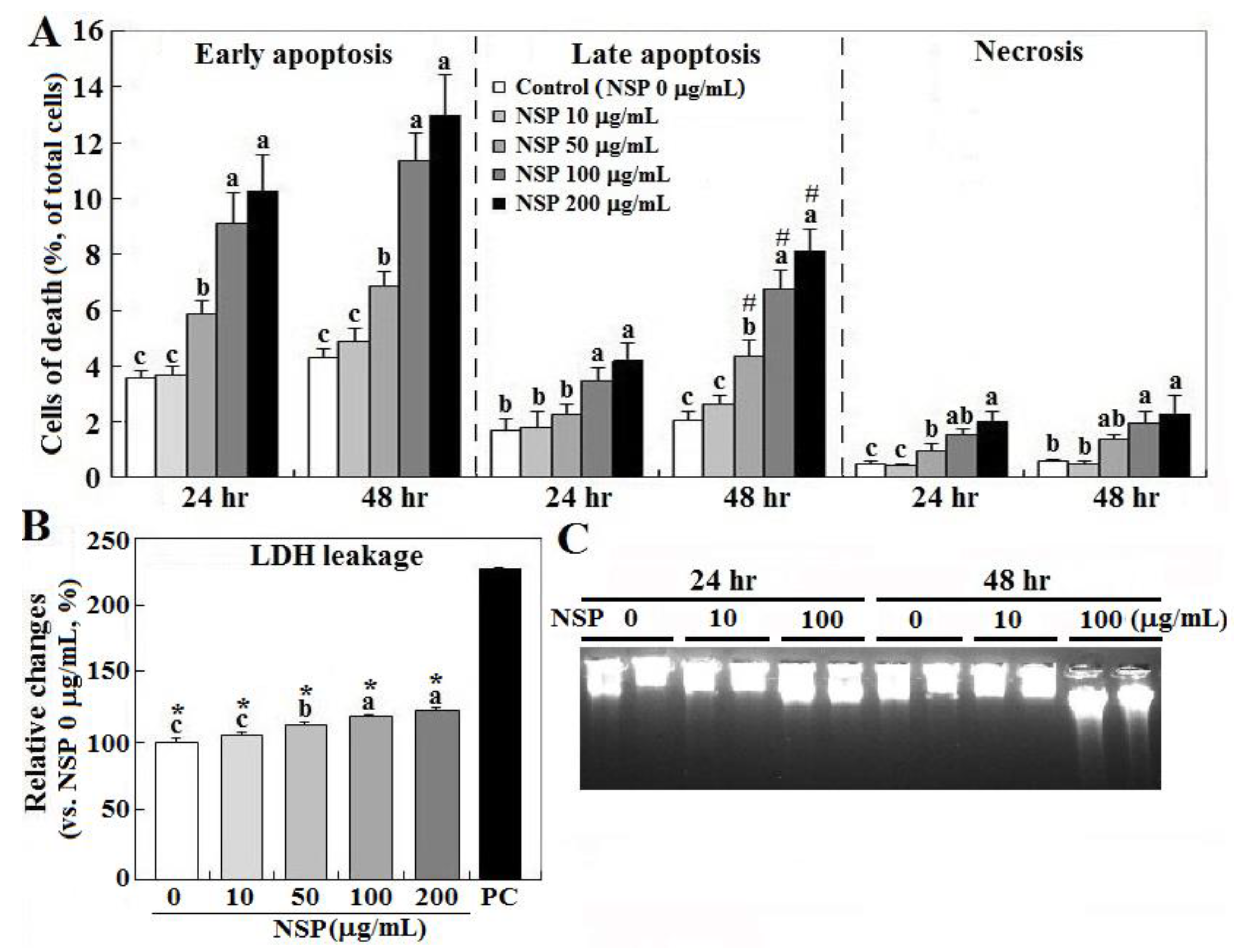{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Cell Death by NSP Exposure
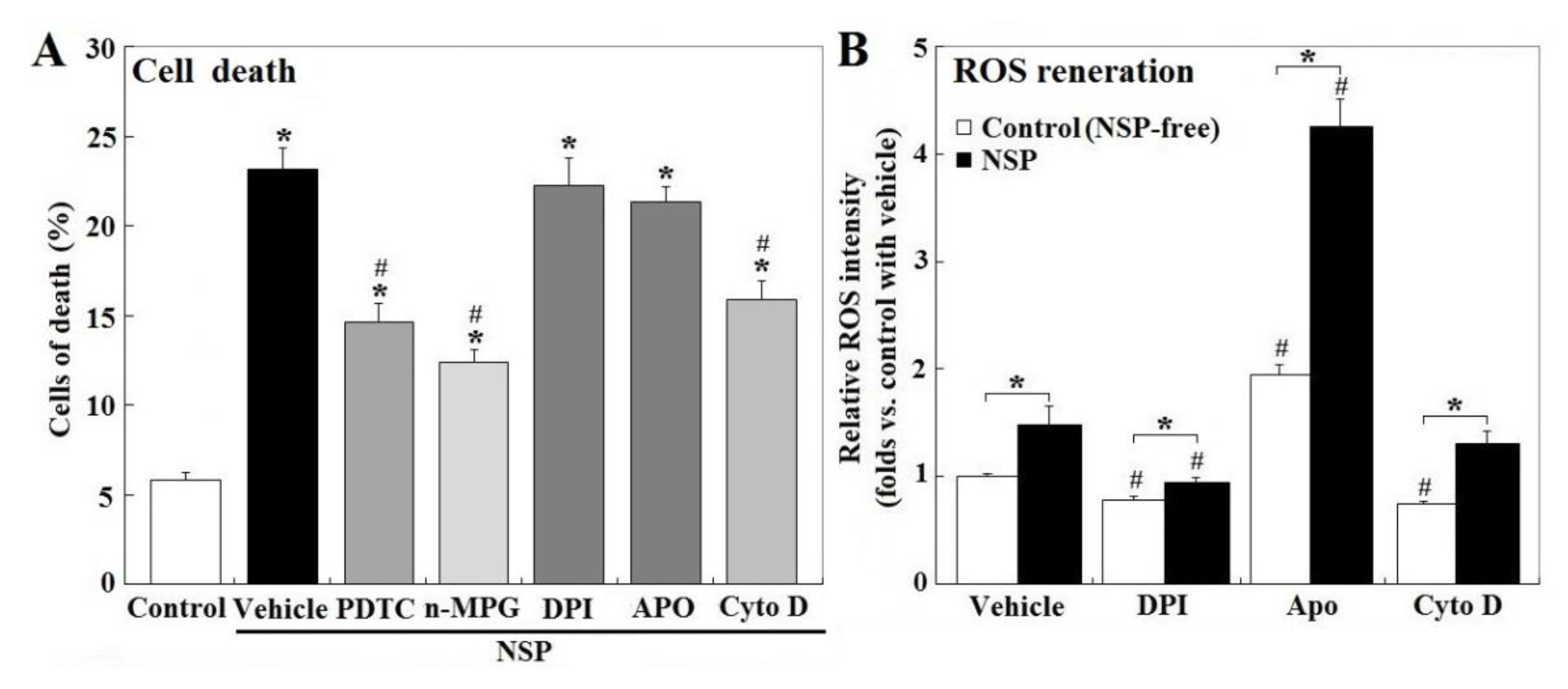
2.2. ROS Contribution and Origins
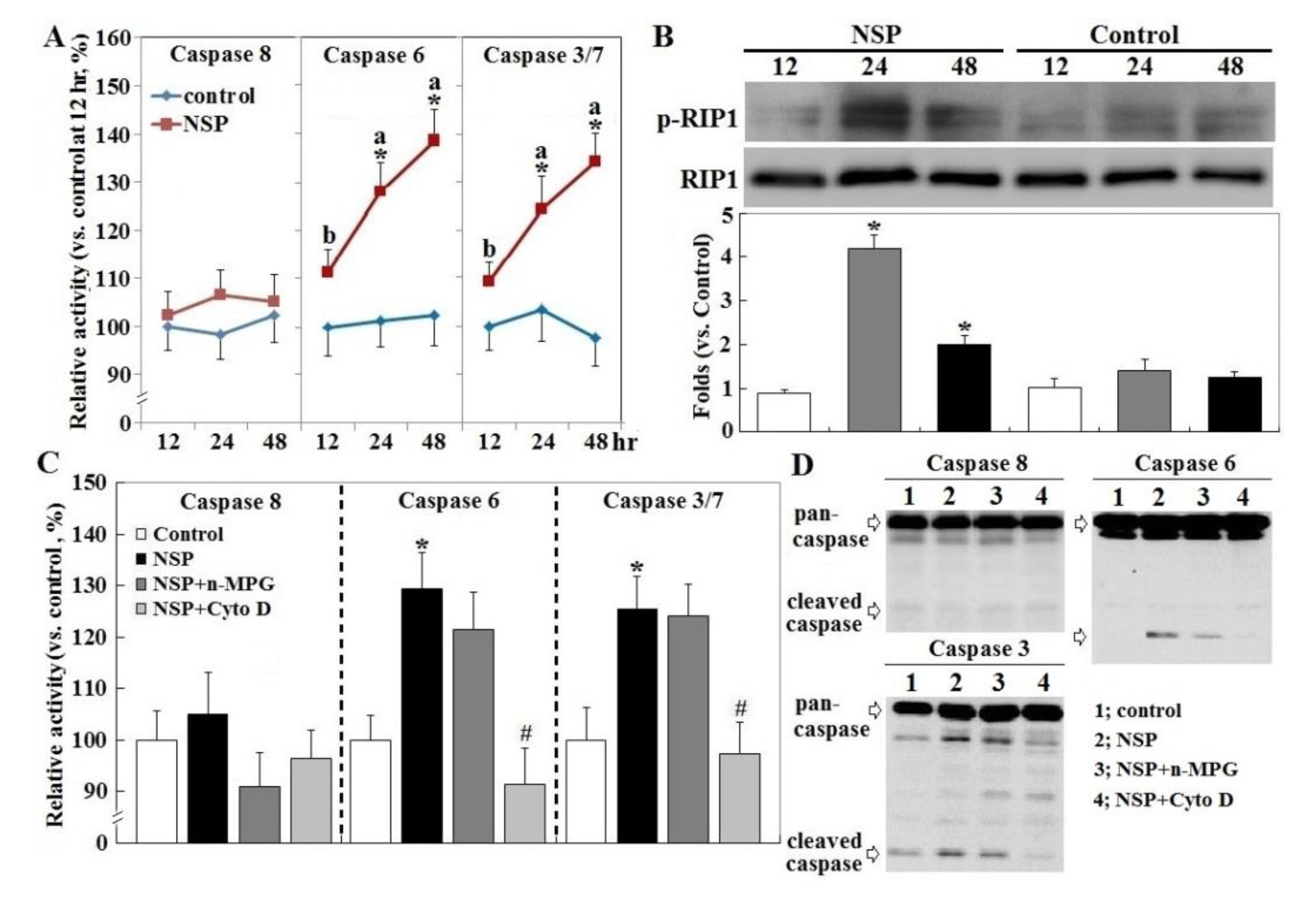
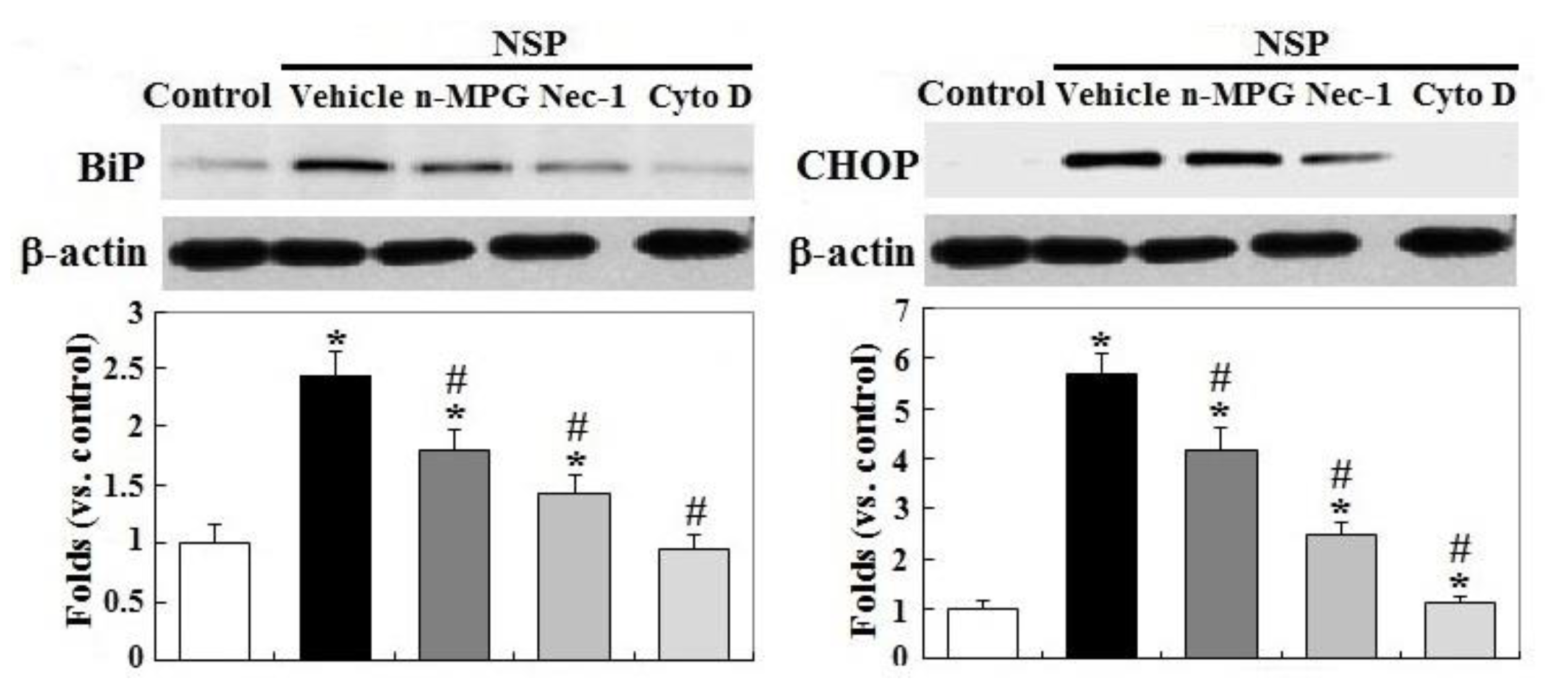
2.3. RIP1 Kinase, Caspase Activation, and ER Stress
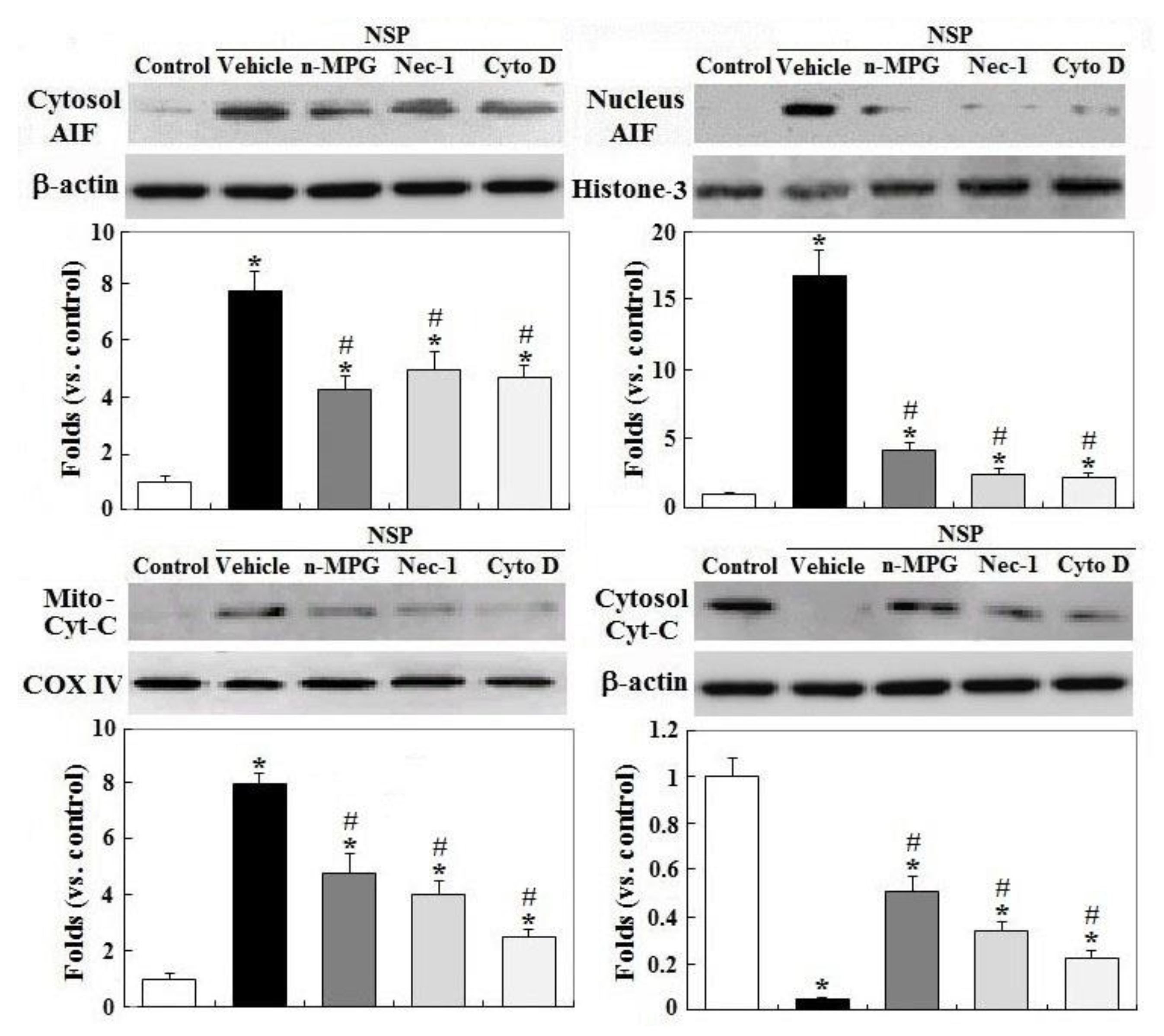
2.4. Mitochondrial Membrane Potential, Cyt-C Release, and AIF Translocation
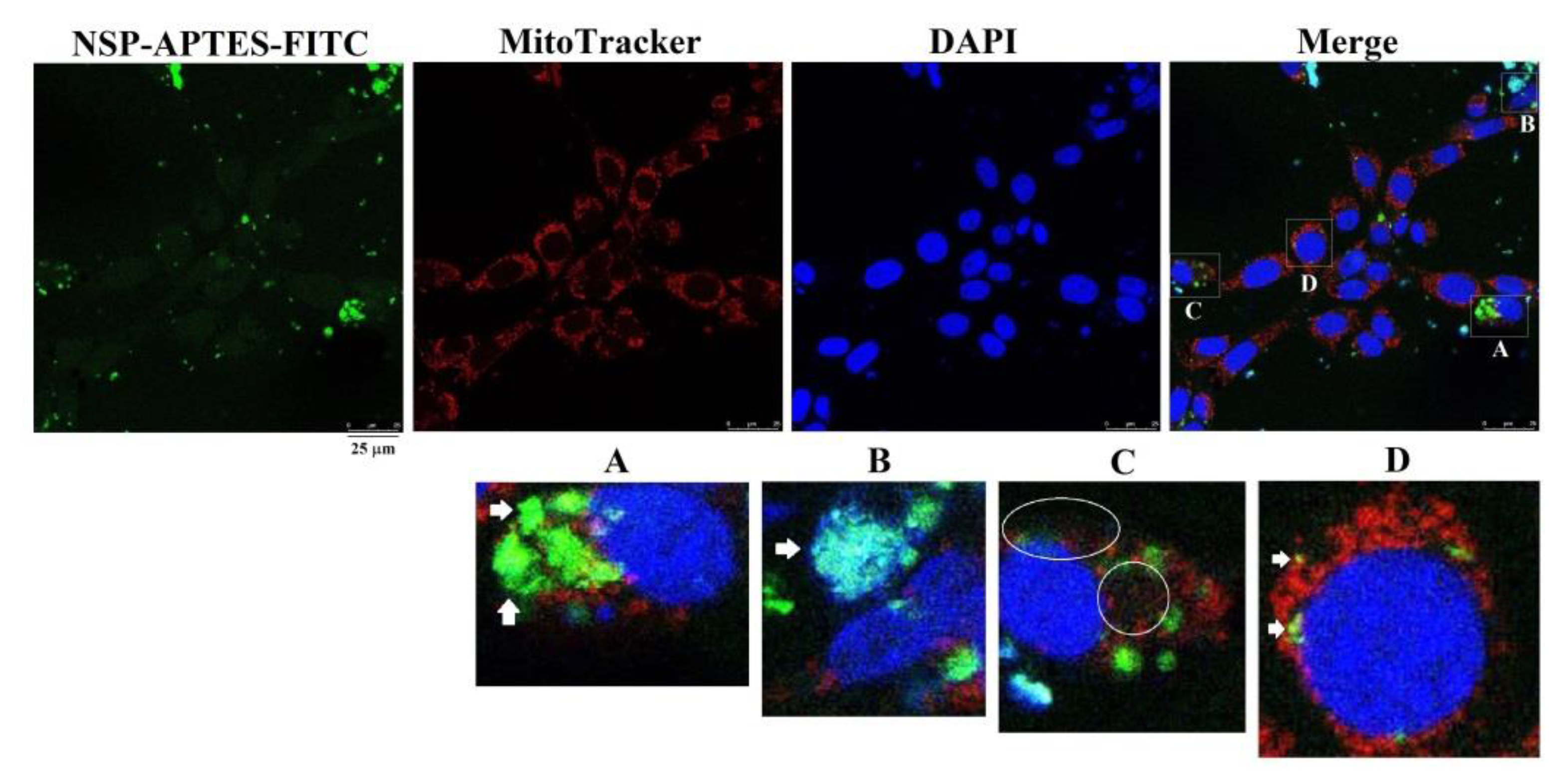
2.5. Localization of NSP Traffic
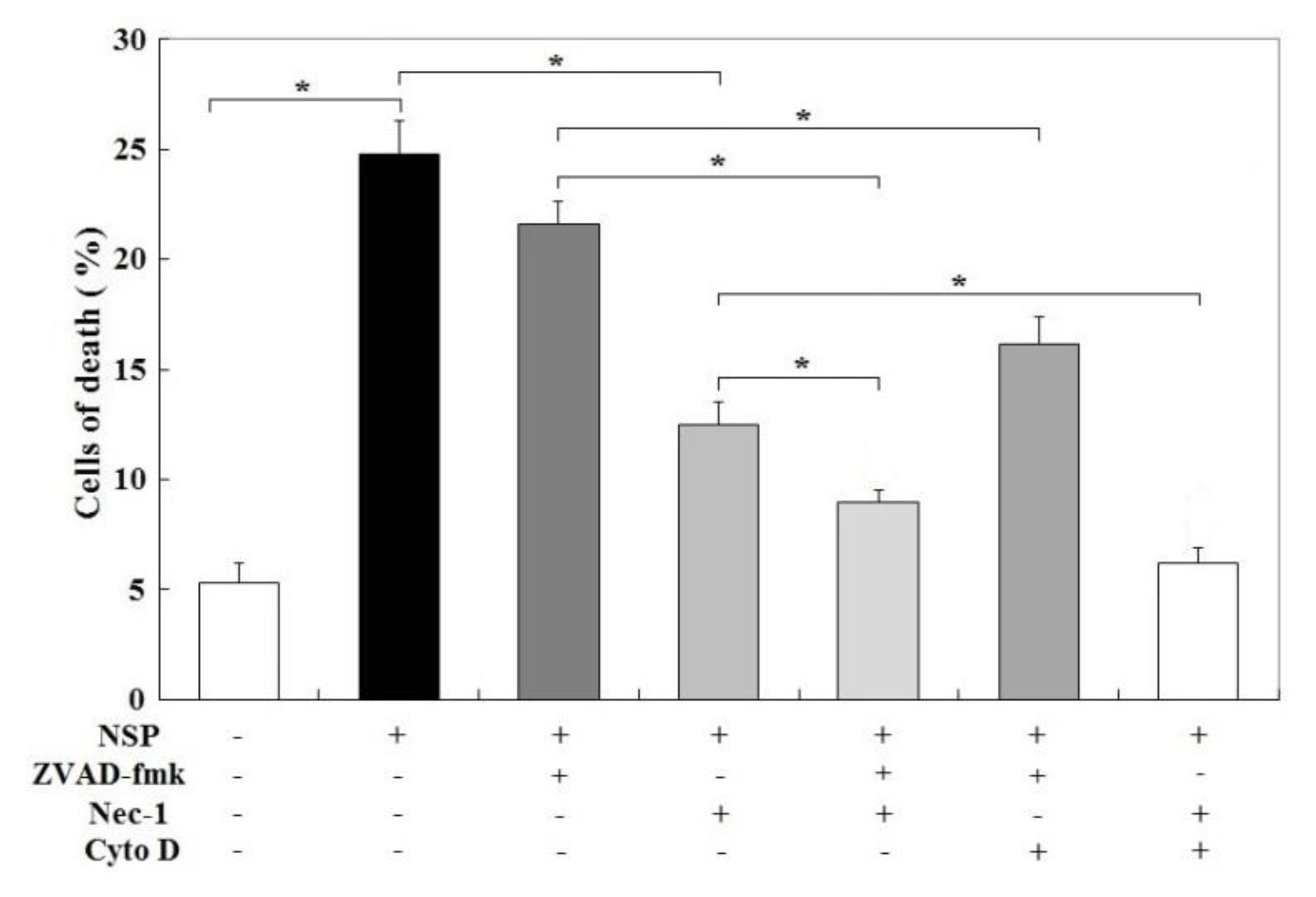
2.6. Caspase and RIP1/3 Kinase Activation, and NSP Internalization in NSP-Induced Cell Death
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Preparations of NSP
5.2. Cell Cultures
5.3. Cell Death and LDH Leakage Analysis
5.4. ROS Production, Mitochondrial Membrane Potential, and Caspase Activity
5.5. Western Blot Analysis
5.6. Subcellular Compartmentalization of NSP
5.7. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gelineau-van Waes, J.; Starr, L.; Maddox, J.; Aleman, F.; Voss, K.A.; Wilberding, J.; Riley, R.T. Maternal fumonisin exposure and risk for neural tube defects: Mechanisms in an in vivo mouse model. Birth Defects Res. Part. Clin. Mol. Teratol. 2005, 73, 487–497. [Google Scholar] [CrossRef]
- Galvano, F.; Piva, A.; Ritieni, A.; Galvano, G.J. Dietary strategies to counteract the effects of mycotoxins: A review. Food Prot. 2001, 64, 120–131. [Google Scholar] [CrossRef]
- Huwig, A.; Freimund, S.; Kappeli, O.; Dutler, H. Mycotoxin detoxification of animal feed by different adsorbents. Toxicol. Lett. 2001, 122, 179–188. [Google Scholar] [CrossRef]
- Dixon, J.B.; Kannewischer, I.; Tenorio Arvide, M.G.; Barrientos Velazquez, A.L. Aflatoxin sequestration in animal feeds by quality-labeled smectite clays: An introductory plan. Appl. Clay Sci. 2008, 40, 201–208. [Google Scholar] [CrossRef]
- Phillips, T.D. Dietary clay in the chemoprevention of aflatoxin induced disease. Toxicol. Sci. 1999, 52, 118–126. [Google Scholar] [CrossRef]
- Yuan, C.W.; Huang, J.T.; Chen, C.C.; Tang, P.C.; Huang, J.W.; Lin, J.J.; Huang, S.Y.; Chen, S.E. Evaluation of Efficacy and Toxicity of Exfoliated Silicate Nanoclays as a Feed Additive for Fumonisin Detoxification. J. Agric. Food Chem. 2017, 65, 6564–6571. [Google Scholar] [CrossRef]
- Tzou, Y.M.; Chan, Y.T.; Chen, S.E.; Wang, C.C.; Chiang, P.N.; Teah, H.Y.; Hung, J.T.; Wu, J.J.; Liu, Y.T. Use 3-D tomography to reveal structural modification of bentonite-enriched clay by nonionic surfactants: Application of organo-clay composites to detoxify aflatoxin B1 in chickens. J. Hazard. Mater. 2019, 375, 312–319. [Google Scholar] [CrossRef]
- Liao, Y.J.; Yang, J.R.; Chen, S.E.; Wu, S.J.; Huang, S.Y.; Lin, J.J.; Chen, L.R.; Tang, P.C. Inhibition of fumonisin B1 cytotoxicity by nanosilicate platelets during mouse embryo development. PLoS ONE 2014, 9, e112290. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.C.; Chiang, M.L.; Tsai, C.M.; Lin, J.J. Exfoliation of montmorillonite clay by mannich polyamines with multiple quaternary salts. Macromolecules 2005, 38, 6240–6243. [Google Scholar] [CrossRef]
- Chiu, C.W.; Huang, T.K.; Wang, Y.C.; Alamani, B.G.; Lin, J.J. Intercalation strategies in clay/polymer hybrids. Prog. Polym. Sci. 2014, 39, 443–485. [Google Scholar] [CrossRef]
- Hsu, S.H.; Tseng, H.J.; Hung, H.S.; Wang, M.C.; Hung, C.H.; Li, P.R.; Lin, J.J. Antimicrobial activities and cellular responses to natural silicate clays and derivatives modified by cationic alkylamine salts. ACS Appl. Mater. Interfaces 2009, 11, 2556–2564. [Google Scholar] [CrossRef] [PubMed]
- Li, P.R.; Wei, J.C.; Chiu, Y.F.; Su, H.L.; Peng, F.C.; Lin, J.J. Evaluation on cytotoxicity and genotoxicity of the exfoliated silicate nanoclay. ACS Appl. Mater. Interfaces 2010, 2, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.C.; Yen, Y.T.; Su, H.L.; Lin, J.J. Inhibition of bacterial growth by the exfoliated clays and observation of physical capturing mechanism. J. Phys. Chem. C 2011, 115, 18770–18775. [Google Scholar] [CrossRef]
- Lin, J.J.; Lin, W.C.; Li, S.D.; Lin, C.Y.; Hsu, S.H. Evaluation of the antibacterial activity and biocompatibility for silver nanoparticles immobilized on nanosilicate platelets. ACS Appl. Mater. Interfaces 2013, 5, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Chiao, S.H.; Lin, S.H.; Shen, C.I.; Liao, J.W.; Bau, I.J.; Wei, J.C.; Tseng, L.P.; Hsu, S.H.; Lai, P.S.; Lin, S.Z.; et al. Efficacy and safety of nanohybrids comprising silver nanoparticles and silicate clay for controlling Salmonella infection. Int. J. Nanomed. 2012, 7, 2421–2432. [Google Scholar]
- Su, H.L.; Lin, S.H.; Wei, J.C.; Pao, I.C.; Chiao, S.H.; Huang, C.C.; Lin, S.Z.; Lin, J.J. Novel nanohybrids of silver particles on clay platelets for inhibiting silver-resistant bacteria. PLoS ONE 2011, 6, e21125. [Google Scholar] [CrossRef]
- Liang, J.J.; Wei, J.C.; Lee, Y.L.; Hsu, S.H.; Lin, J.J.; Lin, Y.L. Surfactant-modified nanoclay exhibits an antiviral activity with high potency and broad spectrum. J. Virol. 2014, 88, 4218–4228. [Google Scholar] [CrossRef] [Green Version]
- Ye, S.; Wang, Y.; Jiao, F.; Zhang, H.; Lin, C.; Wu, Y.; Zhang, Q. The role of NADPH oxidase in multi-walled carbon nanotubes-induced oxidative stress and cytotoxicity in human macrophages. J. Nanosci. Nanotechnol. 2011, 11, 3773–3781. [Google Scholar] [CrossRef]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef] [Green Version]
- Formigli, L.; Papucci, L.; Tani, A.; Schiavone, N.; Tempestini, A.; Orlandini, G.E.; Capaccioli, S.; Orlandini, S.Z. Aponecrosis: Morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J. Cell Physiol. 2000, 182, 41–49. [Google Scholar] [CrossRef]
- Kusano, H.; Shimizu, S.; Koya, R.C.; Fujita, H.; Kamada, S.; Kuzumaki, N.; Tsujimoto, Y. Human gelsolin prevents apoptosis by inhibiting apoptotic mitochondrial changes via closing VDAC. Oncogene 2000, 19, 4807–4814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luschen, S.; Ussat, S.; Scherer, G.; Kabelitz, D.; Adam-Klages, S. Sensitization to death receptor cytotoxicity by inhibition of Fas-associated death domain protein (FADD)/caspase signaling. J. Biol. Chem. 2000, 275, 24670–24678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delavallee, L.; Cabon, L.; Galan-Malo, P.; Lorenzo, H.K.; Susin, S.A. AIF-mediated caspase-independent necroptosis: A new chance for targeted therapeutics. IUBMB Life 2011, 63, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Lordan, S.; Kennedy, J.E.; Higginbotham, C.L. Cytotoxic effects induced by unmodified and organically modified nanoclays in the human hepatic HepG2 cell line. J. Appl. Toxicol. 2011, 31, 27–35. [Google Scholar] [CrossRef]
- Nel, A.; Xia, T.; Madler, L.; Li, N. Toxic potential of materials at the nanolevel. Science 2006, 311, 622–627. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Miller, J.W.; Vavvas, D.G. RIP kinase-mediated necrosis as an alternative mechanisms of photoreceptor death. Oncotarget 2011, 2, 497–509. [Google Scholar] [CrossRef] [Green Version]
- Luders, J.; Demand, J.; Höhfeld, J. The ubiquitin-related BAG-1 provides a link between the molecular chaperones Hsc70/Hsp70 and the proteasome. J. Biol. Chem. 2000, 275, 4613–4617. [Google Scholar] [CrossRef] [Green Version]
- Egger, L.; Schneider, L.; Rheme, C.; Tapernoux, M.; Hacki, J.; Borner, C. Serine proteases mediate apoptosis-like cell death and phagocytosis under caspase-inhibiting conditions. Cell Death Differ. 2003, 10, 1188–1203. [Google Scholar] [CrossRef] [Green Version]
- Nabeshi, H.; Yoshikawa, T.; Matsuyama, K.; Nakazato, Y.; Tochigi, S.; Kondoh, S.; Hirai, T.; Akase, T.; Nagano, K.; Abe, Y.; et al. Amorphous nanosilica induce endocytosis-dependent ROS generation and DNA damage in human keratinocytes. Part. Fibre Toxicol. 2011, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Frohlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [Green Version]
- Janer, G.; Fernandez-Rosas, E.; Mas del Molino, E.; Gonzalez-Galvez, D.; Vilar, G.; Lopez-Iglesias, C.; Ermini, V.; Vazquez-Campos, S. In vitro toxicity of functionalized nanoclays is mainly driven by the presence of organic modifiers. Nanotoxicology 2014, 8, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.L.; Pan, Y.E.; Chang, C.J.; Tang, P.C.; Huang, Y.F.; Walzem, R.L.; Chen, S.E. Palmitic acid in chicken granulosa cell death-lipotoxic mechanisms mediate reproductive inefficacy of broiler breeder hens. Theriogenology 2012, 78, 1917–1928. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.C.; Xie, Y.L.; Chang, C.J.; Su, C.M.; Chen, Y.H.; Huang, S.Y.; Walzem, R.L.; Chen, S.E. Feed intake alters immune cell functions and ovarian infiltration in broiler hens-implications for reproductive performance. Biol. Reprod. 2014, 90, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rottenberg, H.; Wu, S. Quantitative assay by flow cytometry of the mitochondrial membrane potential in intact cells. Biochim. Biophys. Acta 1998, 1404, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.C.; Chiang, S.K.; Chen, S.E.; Yu, Y.L.; Chou, R.H.; Chang, W.C. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018, 416, 124–137. [Google Scholar] [CrossRef]
- Lin, J.-J.; Chang, C.-H.; Lee, B.-H.; Liao, Q.-Y.; Chen, J.-J. Modified Nano Silicate Platelet and Application of Modified Nano silicate Platelet for Preparing Cancer Treating Medicine. ROC Patent I681,777, 16 March 2019. [Google Scholar]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.-T.; Chang, L.-C.; Cheng, C.-S.; Lin, J.-J.; Huang, S.-Y.; Chen, S.-E. Cytotoxicity Produced by Silicate Nanoplatelets: Study of Cell Death Mechanisms. Toxins 2020, 12, 623. https://doi.org/10.3390/toxins12100623
Huang J-T, Chang L-C, Cheng C-S, Lin J-J, Huang S-Y, Chen S-E. Cytotoxicity Produced by Silicate Nanoplatelets: Study of Cell Death Mechanisms. Toxins. 2020; 12(10):623. https://doi.org/10.3390/toxins12100623
Chicago/Turabian StyleHuang, Jie-Ting, Ling-Chu Chang, Chung-Ssu Cheng, Jiang-Jen Lin, San-Yuan Huang, and Shuen-Ei Chen. 2020. "Cytotoxicity Produced by Silicate Nanoplatelets: Study of Cell Death Mechanisms" Toxins 12, no. 10: 623. https://doi.org/10.3390/toxins12100623
APA StyleHuang, J. -T., Chang, L. -C., Cheng, C. -S., Lin, J. -J., Huang, S. -Y., & Chen, S. -E. (2020). Cytotoxicity Produced by Silicate Nanoplatelets: Study of Cell Death Mechanisms. Toxins, 12(10), 623. https://doi.org/10.3390/toxins12100623