Should We Consider the Cardiovascular System While Evaluating CKD-MBD?


Abstract
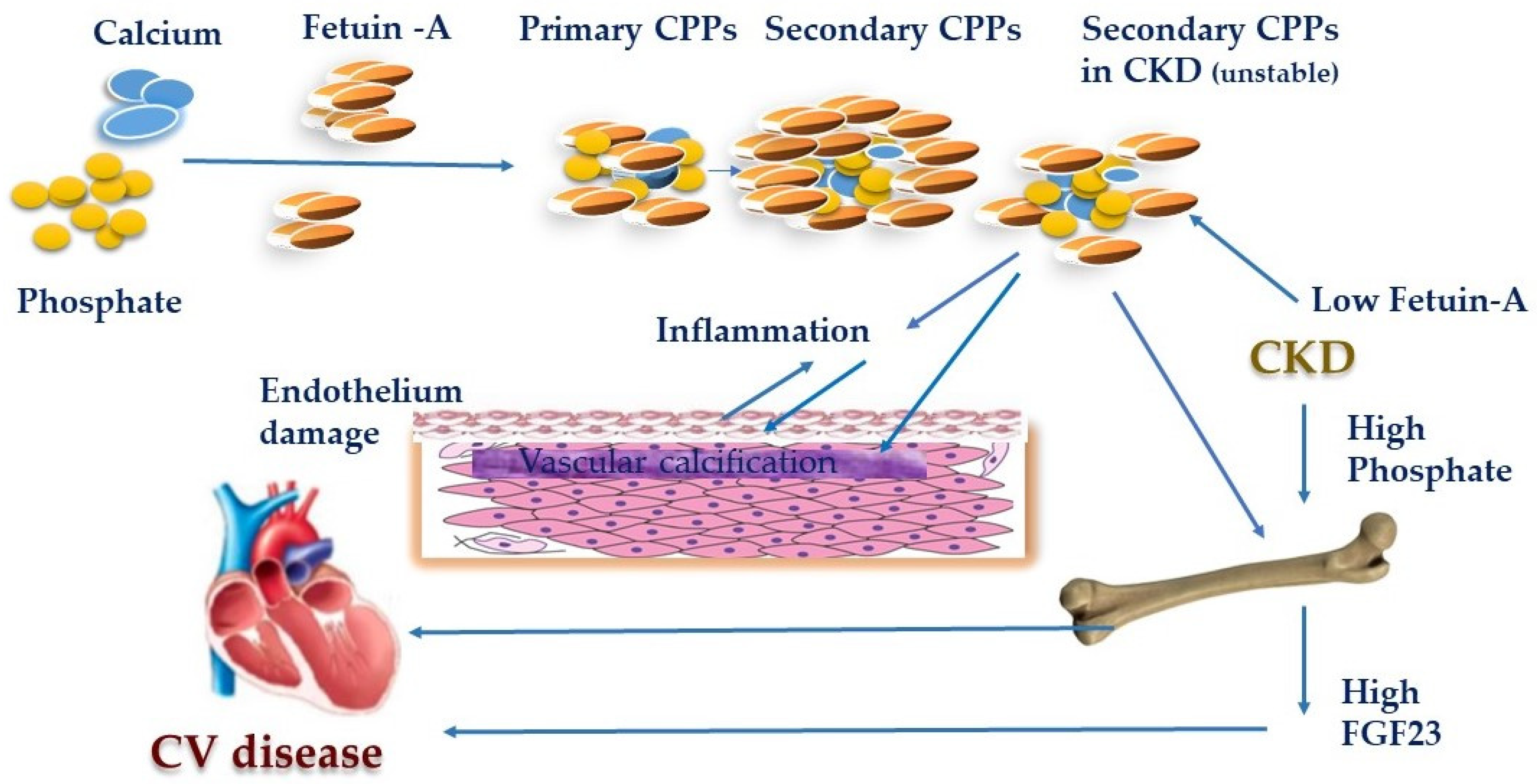
:1. Introduction
2. Role of Phosphate, Parathyroid Hormone and Vit D Deficiency in Uremic Cardiomyopathy
2.1. Pathophysiology of Uremic Cardiomyopathy in CKD Patients
2.1.1. Left Ventricular Cardiomyopathy
2.1.2. Interstitial Fibrosis
2.1.3. Microvascular Disease
2.2. Role of Phosphate in Uremic Cardiomyopathy
2.3. Role of Parathyroid Hormone
2.4. Role of Vitamin D
3. Importance of New CKD-MBD Biomarkers in Early Cardiovascular Risk Assessment
3.1. Role of FGF23
3.2. Role of Klotho
3.3. Role of Sclerostin
3.4. Role of OPG-RANK-RANKL System in CKD-MBD
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States; National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2013; Available online: www.usrds.org/atlas (accessed on 21 February 2020).
- Gargiulo, R.; Suhail, F.; Lerma, E. Cardiovascular disease and chronic kidney disease. Dis Mon. 2015, 61, 403–413. [Google Scholar] [CrossRef] [PubMed]
- De Albuquerque Suassuna, P.G.; Sanders-Pinheiro, H.; De Paula, R.B. Uremic Cardiomyopathy: A New Piece in the Chronic Kidney Disease-Mineral and Bone Disorder Puzzle. Front. Med. 2018, 5, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remppis, A.; Ritz, E. Cardiac problems in the dialysis patient: Beyond coronary disease. Semin. Dial. 2008, 21, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Di Lullo, L.; Gorini, A.; Russo, D.; Santoboni, A.; Ronco, C. Left Ventricular Hypertrophy in Chronic Kidney Disease Patients: From Pathophysiology to Treatment. Cardio Renal. Med. 2015, 5, 254–266. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Shapiro, J.I. Evolving concepts in the pathogenesis of uraemic cardiomyopathy. Nat. Rev. Nephrol. 2019. [Google Scholar] [CrossRef]
- Hruska, K.A.; Seife, M.; Sugatani, T. Pathophysiology of the Chronic Kidney Disease—Mineral Bone Disorder (CKD-MBD). Curr. Opin. Nephrol. Hypertens. 2015, 24, 303–309. [Google Scholar]
- D’Marco, L.; Bellasi, A.; Raggi, P. Cardiovascular biomarkers in chronic kidney disease: State of current research and clinical applicability. Dis. Markers 2015. [Google Scholar] [CrossRef] [Green Version]
- Amann, K.; Breitbach, M.; Ritz, E.; Mall, G. Myocyte/capillary mismatch in the heart of uremic patients. J. Am. Soc. Nephrol. 1998, 9, 1018–1022. [Google Scholar]
- Chinnappa, S.; Hothi, S.S.; Tan, L.B. Is uremic cardiomyopathy a direct consequence of chronic kidney disease? Expert Rev. Cardiovasc. Ther. 2014, 12, 127–130. [Google Scholar] [CrossRef]
- Chirakarnjanakorn, S.; Navaneethan, S.D.; Francis, G.S.; Tang, W.H. Cardiovascular impact in patients undergoing maintenance hemodialysis: Clinical management considerations. Int. J. Cardiol. 2017, 232, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Grabner, A.; Faul, C. The Role of FGF23 and Klotho in Uremic Cardiomyopathy. Curr. Opin. Nephrol. Hypertens 2016, 25, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.L.; Ritz, E. Hypertrophy and fibrosis in the cardiomyo pathy of uremia—Beyond coronary heart disease. Semin. Dial. 2008, 21, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Ritz, E. Left ventricular hypertrophy in renal disease: Beyond preload and afterload. Kidney Int. 2009, 75, 771–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedecostante, M.; Spannella, F.; Cola, G.; Espinosa, E.; Dessì-Fulgheri, P.; Sarzani, R. Chronic kidney disease is characterized by “double trouble” higher pulse pressure plus night-time systolic blood pressure and more severe cardiac damage. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Viegas, C.; Araújo, N.; Marreiros, C.; Simes, D. The interplay between mineral metabolism, vascular calcification and inflammation in Chronic Kidney Disease (CKD): Challenging old concepts with new facts. Aging 2019, 11, 4274–4299. [Google Scholar] [CrossRef]
- Valdivielso, J.M.; Rodríguez-Puyol, D.; Pascua, J.; Barrios, C.; Bermúdez-López, M.; Sánchez-Niño, M.D.; Pérez-Fernández, M.; Ortiz, A. Atherosclerosis in Chronic Kidney Disease: More, Less, or Just Different? Arterioscler Thromb. Vasc. Biol. 2019, 39, 1938–1966. [Google Scholar] [CrossRef]
- Alhaj, E.; Alhaj, N.; Rahman, I.; Niazi, T.O.; Berkowitz, R.; Klapholz, M. Uremic Cardiomyopathy: An Underdiagnosed Disease. Congest. Heart Fail. 2013, 19, 40–45. [Google Scholar] [CrossRef]
- Ikram, H.; Lynn, K.L.; Bailey, R.R.; Little, P.J. Cardiovascular changes in chronic hemodialysis patients. Kidney Int. 1983, 24, 371–376. [Google Scholar] [CrossRef] [Green Version]
- Mall, G.; Huther, W.; Schneider, J.; Lundin, P.; Ritz, E. Diffuse intermyocardiocytic fibrosis in uraemic patients. Nephrol. Dial. Transpl. 1990, 5, 39–44. [Google Scholar] [CrossRef]
- Mall, G.; Rambausek, M.; Neumeister, A.; Kollmar, S.; Vetterlein, F.; Ritz, E. Myocardial interstitial fibrosis in experimental uremia--implications for cardiac compliance. Kidney Int. 1988, 33, 804–811. [Google Scholar] [CrossRef] [Green Version]
- Hayer, M.K.; Price, A.M.; Liu, B.; Baig, S.; Ferro, C.J.; Townend, J.N.; Steeds, R.P.; Edwards, N.C. Diffuse Myocardial Interstitial Fibrosis and Dysfunction in Early Chronic Kidney Disease. Am. J. Cardiol. 2018, 121, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoccali, C.; Benedetto, F.A.; Tripepi, G.; Mallamaci, F. Cardiac consequences of hypertension in hemodialysis patients. Semin. Dial. 2004, 17, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Rostand, S.G.; Kirk, K.A.; Rutsky, E.A. The epidemiology of coronary artery disease in patients on maintenance hemodialysis: Implications for management. Contrib. Nephrol. 1986, 52, 34–41. [Google Scholar] [PubMed]
- Mohandas, R.; Segal, M.S.; Huo, T.; Handberg, E.M.; Petersen, J.W.; Johnson, B.D.; Pepine, C.J. Renal Function and Coronary Microvascular Dysfunction in Women with Symptoms/Signs of Ischemia. PLoS ONE 2015, 10, e0125374. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, U.; Buzello, M.; Ritz, E.; Stein, G.; Raabe, G.; Wiest, G.; Mall, G.; Amann, K. Morphology of coronary atherosclerotic lesions in patients with end-stage renal failure. Nephrol. Dial. Transpl. 2000, 15, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Colbert, G.; Jain, N.; De Lemos, J.A.; Hedayati, S.S. Utility of traditional circulating and imaging-based cardiac biomarkers in patients with predialysis CKD. Clin. J. Am. Soc. Nephrol. 2015, 10, 515–529. [Google Scholar] [CrossRef] [Green Version]
- Morena, M.; Jaussent, I.; Dupuy, A.M.; Bargnoux, A.S.; Kuster, N.; Chenine, L.; Leray-Moragues, H.; Klouche, K.; Vernhet, H.; Canaud, B.; et al. Osteoprotegerin and sclerostin in chronic kidney disease prior to dialysis: Potential partners in vascular calcifications. Nephrol. Dial. Transpl. 2015, 30, 1345–1356. [Google Scholar] [CrossRef] [Green Version]
- Lutsey, P.L.; Alonso, A.; Michos, E.D.; Loehr, L.R.; Astor, B.C.; Coresh, J.; Folsom, A.R. Serum magnesium, phosphorus, and calcium are associated with risk of incident heart failure: The Atherosclerosis Risk in Communities (ARIC) Study. Am. J. Clin. Nutr. 2014, 100, 756–764. [Google Scholar] [CrossRef]
- Vervloet, M.G.; Massy, Z.A.; Brandenburg, V.M.; Mazzaferro, S.; Cozzolino, M.; Ureña-Torres, P.; Bover, J.; Goldsmith, D. CKD-MBD Working Group of ERA-EDTA. Bone: A new endocrine organ at the heart of chronic kidney disease and mineral and bone disorders. Lancet Diabetes Endocrinol. 2014, 2, 427–436. [Google Scholar] [CrossRef]
- Vervloet, M. Modifying Phosphate Toxicity in Chronic Kidney Disease. Toxins 2019, 11, 522. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.C.; Shiizaki, K.; Kuro-O, M.; Moe, O.W. Fibroblast growth factor 23 and Klotho: Physiology and pathophysiology of an endocrine network of mineral metabolism. Annu. Rev. Physiol. 2013, 75, 503–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Paloian, N.J.; Giachelli, C.M. A current understanding of vascular calcification in CKD. Am. J. Physiol. Renal. Physiol. 2014, 307, 891–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giachelli, C.M. The emerging role of phosphate in vascular calcification. Kidney Int. 2009, 75, 890–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razzaque, M.S. Phosphate Toxicity and Vascular Mineralization. Phosphate and Vitamin D in Chronic Kidney Disease. Contrib. Nephrol. 2013, 180, 74–85. [Google Scholar]
- Taniguchi, M.; Fukagawa, M.; Fujii, N.; Hamano, T.; Shoji, T.; Yokoyama, K.; Nakai, S.; Shigematsu, T.; Iseki, K.; Tsubakihara, Y. Serum phosphate and calcium should be primarily and consistently controlled in prevalent hemodialysis patients. Ther. Apher. Dial. 2013, 17, 221–228. [Google Scholar] [CrossRef]
- Rroji, M.; Seferi, S.; Cafka, M.; Petrela, E.; Likaj, E.; Barbullushi, M.; Thereska, N.; Spasovski, G. Is residual renal function and better phosphate control in peritoneal dialysis an answer for the lower prevalence of valve calcification compared to hemodialysis patients? Int. Urol. Nephrol. 2014, 46, 175–182. [Google Scholar] [CrossRef]
- Fujii, H.; Joki, N. Mineral metabolism and cardiovascular disease in CKD. Clin. Exp. Nephrol. 2017, 21, 53–63. [Google Scholar] [CrossRef]
- Peng, A.; Wu, T.; Zeng, C.; Rakheja, D.; Zhu, J.; Ye, T.; Hutcheson, J.; Vaziri, N.D.; Liu, Z.; Mohan, C.; et al. Adverse effects of simulated hyper- and hypo-phosphatemia on endothelial cell function and viability. PLoS ONE 2011, 6, e23268. [Google Scholar] [CrossRef] [Green Version]
- Di Marco, G.S.; Hausberg, M.; Hillebrand, U.; Rustemeyer, P.; Wittkowski, W.; Lang, D.; Pavenstädt, H. Increased inorganic phosphate induces human endothelial cell apoptosis in vitro. Am. J. Physiol. Renal. Physiol. 2008, 294, 1381–1387. [Google Scholar] [CrossRef]
- Di Marco, G.S.; König, M.; Stock, C.; Wiesinger, A.; Hillebrand, U.; Reiermann, S.; Reuter, S.; Amler, S.; Köhler, G.; Buck, F.; et al. High phosphate directly affects endothelial function by downregulating annexin II. Kidney Int. 2013, 83, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Koc, M.; Bihorac, A.; Segal, M.S. Circulating endothelial cells as potential markers of the state of the endothelium in hemodialysis patients. Am. J. Kidney Dis. 2003, 42, 704–712. [Google Scholar] [CrossRef]
- Kuro-O, M. Calciprotein particle (CPP): A true culprit of phosphorus woes? Nefrologia 2014, 34, 1–4. [Google Scholar] [PubMed]
- Akiyama, K. Calciprotein particle contributes to the synthesis and secretion of fibroblast growth factor 23 induced by dietary phosphate intake. J. Am. Soc. Nephrol. 2017, 28, 210. [Google Scholar]
- Viegas, C.S.B.; Santos, L.; Macedo, A.L.; Matos, A.A.; Silva, A.P.; Neves, P.L.; Staes, A.; Gevaert, K.; Morais, R.; Vermeer, C.; et al. Chronic Kidney Disease Circulating Calciprotein Particles and Extracellular Vesicles Promote Vascular Calcification: A Role for GRP (Gla-Rich Protein). Arterioscler Thromb. Vasc. Biol. 2018, 38, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, K.; Kimura, T.; Shiizaki, K. Biological and Clinical Effects of Calciprotein Particles on Chronic Kidney Disease-Mineral and Bone Disorder. Int. J. Endocrinol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Ciceri, P.; Falleni, M.; Tosi, D.; Martinelli, C.; Cannizzo, S.; Bulfamante, G.; Block, G.A.; Marchetti, G.; Cozzolino, M. Therapeutic Effect of Iron Citrate in Blocking Calcium Deposition in High Pi-Calcified VSMC: Role of Autophagy and Apoptosis. Int. J. Mol. Sci. 2019, 20, 5925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han-Kyul, K.; Masaki, M.; Wanpen, V. Phosphate, the forgotten mineral in hypertension. Curr. Opin. Nephrol. Hypertens. 2019, 28, 345–351. [Google Scholar]
- Amann, K.; Törnig, J.; Kugel, B.; Gross, M.L.; Tyralla, K.; El-Shakmak, A.; Szabo, A.; Ritz, E. Hyperphosphatemia aggravates cardiac fibrosis and microvascular disease in experimental uremia. Kidney Int. 2003, 63, 1296–1301. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.C.; Shi, M.; Cho, H.J.; Adams-Huet, B.; Paek, J.; Hill, K.; Shelton, J.; Amaral, A.P.; Faul, C.; Taniguchi, M.; et al. Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J. Am. Soc. Nephrol. 2015, 26, 1290–1302. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Qin, L.; Wu, T.; Deng, B.; Sun, Y.; Hu, D.; Mohan, C.; Zhou, X.J.; Peng, A.L. Elevated Cardiac Markers in Chronic Kidney Disease as a Consequence of Hyperphosphatemia-Induced Cardiac Myocyte Injury. Med. Sci. Monit. 2014, 20, 2043–2053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covic, A.; Kothawala, P.; Nernal, M.; Robbins, S.; Chalian, A.; Goldsmith, D. Systematic review of the evidence underlying the association between mineral metabolism disturbances and risk of all-cause mortality, cardiovascular mortality and cardiovascular events in chronic kidney disease. Nephrol. Dial. Transpl. 2009, 24, 1506–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomaschitz, A.; Ritz, E.; Pieske, B.; Rus-Machan, J.; Kienreich, K.; Verhyen, N.; Gaksch, M.; Gruber, M.; Fahrleitner-Pammer, A.; Mrak, P.; et al. Aldosterone and parathyroid hormone interactions as mediators of metabolic and cardiovascular disease. Metabolism 2014, 63, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Bogin, E.; Massry, S.G.; Harary, I. Effect of parathyroid-hormone on rat heart cells. J. Clin. Investig. 1981, 67, 1215–1227. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.; Rodriguez, M.; Slatopolsky, E. FGF23 and PTH—Double agents at the heart of CKD. Nephrol. Dial. Transpl. 2012, 27, 1715–1720. [Google Scholar] [CrossRef] [PubMed]
- Neves, K.R.; Graciolli, F.G.; Dos Reis, L.M.; Pasqualucci, C.A.; Moysés, R.M.; Jorgetti, V. Adverse effects of hyperphosphatemia on myocardial hypertrophy, renal function, and bone in rats with renal failure. Kidney Int. 2004, 66, 2237–2244. [Google Scholar] [CrossRef] [Green Version]
- Tomaschitz, A.; Ritz, E.; Pieske, B.; Fahrleitner-Pammer, A.; Kienreich, K.; Horina, J.H.; Drechsler, C.; März, W.; Ofner, M.; Pieber, R.; et al. Aldosterone and parathyroid hormone: A precarious couple for cardiovascular disease. Cardiovasc. Res. 2012, 94, 10–19. [Google Scholar] [CrossRef]
- Schluter, K.D.; Piper, H.M. Trophic effects of catecholamines and parathyroid hormone on adult ventricular cardiomyocytes. Am. J. Physiol. 1992, 263, 1739–1746. [Google Scholar] [CrossRef]
- Custódio, M.R.; Koike, M.K.; Neves, K.R.; Dos Reis, L.M.; Graciolli, F.G.; Neves, C.L.; Batista, D.G.; Magalhães, A.O.; Hawlitschek, P.; Oliveira, I.B.; et al. Parathyroid hormone and phosphorus overload in uremia: Impact on cardiovascular system. Nephrol. Dial. Transpl. 2012, 27, 1437–1445. [Google Scholar] [CrossRef] [Green Version]
- Palmeri, N.O.; Walker, M.D. Parathyroid Hormone and Cardiac Electrophysiology: A Review. Cardiol. Rev. 2019, 27, 182–188. [Google Scholar] [CrossRef]
- Potthoff, S.A.; Janus, A.; Hoch, H.; Frahnert, M.; Tossios, P.; Reber, D.; Giessing, M.; Klein, H.M.; Schwertfeger, E.; Quack, I.; et al. PTH-receptors regulate norepinephrine release in human heart and kidney. Regul. Pept. 2011, 171, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Drüeke, T.; Fauchet, M.; Fleury, J.; Lesourd, P.; Toure, Y.; Le Pailleur, C.; De Vernejoul, P.; Crosnier, J. Effect of parathyroidectomy on left-ventricular function in haemodialysis patients. Lancet 1980, 1, 112–114. [Google Scholar] [CrossRef]
- London, G.M.; Fabiani, F.; Marchais, S.J.; De Vernejoul, M.C.; Guerin, A.P.; Safar, M.E.; Metivier, F.; Llach, F. Uremic cardiomyopathy: An inadequate left ventricular hypertrophy. Kidney Int. 1987, 31, 973–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coratelli, P.; Buongiorno, E.; Petrarulo, F.; Corciulo, R.; Giannattasio, M.; Passavanti, G.; Antonelli, G. Pathogenetic aspects of uremic cardiomyopathy. Miner Electrolyte Metab. 1989, 15, 246–253. [Google Scholar]
- Fellner, S.K.; Lang, R.M.; Neumann, A.; Bushinsky, D.A.; Borow, K.M. Parathyroid hormone and myocardial performance in dialysis patients. Am. J. Kidney Dis. 1991, 18, 320–325. [Google Scholar] [CrossRef]
- Evolve Trial Investigators; Chertow, G.M.; Block, G.A.; Correa-Rotter, R.; Drüeke, T.B.; Floege, J.; Goodman, W.G.; Herzog, C.A.; Kubo, Y.; London, G.M.; et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N. Engl. J. Med. 2012, 367, 2482–2494. [Google Scholar]
- Jorde, R.; Svartberg, J.; Sundsfjord, J. Serum parathyroid hormone as a predictor of increase in systolic blood pressure in men. J. Hypertens. 2005, 23, 1639–1644. [Google Scholar] [CrossRef] [Green Version]
- Pascale, A.V.; Inelli, R.; Giannotti, R.; Visco, V.; Fabbricatore, D.; Matula, I.; Mazzeo, P.; Ragosa, N.; Massari, A.; Izzo, R.; et al. Vitamin D, parathyroid hormone and cardiovascular risk: The good, the bad and the ugly. J. Cardiovasc. Med. 2018, 19, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Noce, A.; Canale, M.P.; Capria, A.; Rovella, V.; Tesauro, M.; Splendiani, G.; Annicchiarico-Petruzzelli, M.; Manzuoli, M.; Simonetti, G.; Di Daniele, N.; et al. Coronary artery calcifications predict long term cardiovascular events in nondiabetic Caucasian hemodialysis patients. Aging 2015, 7, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Baigent, C.; Landray, M.J.; Reith, C.; Emberson, J.; Wheeler, D.C.; Tomson, C.; Wanner, C.; Krane, V.; Cass, A.; Craig, J.; et al. SHARP Investigators: The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (study of heart and renal protection): A randomized placebo-controlled trial. Lancet 2011, 377, 2181–2192. [Google Scholar] [CrossRef] [Green Version]
- Schlieper, G.; Schurgers, L.; Brandenburg, V.; Reutelingsperger, C.; Floege, J. Vascular calcification in chronic kidney disease: An update. Nephrol. Dial. Transpl. 2016, 31, 31–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stam, F.; Van Guldener, C.; Becker, A.; Dekker, J.M.; Heine, R.J.; Bouter, L.M.; Stehouwer, C.D. Endothelial dysfunction contributes to renal function associated cardiovascular mortality in a population with mild renal insufficiency: The Hoorn study. J. Am. Soc. Nephrol. 2006, 17, 537–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vimaleswaran, K.S.; Cavadino, A.; Berry, D.J.; Jorde, R.; Dieffenbach, A.K.; Lu, C.; Jorde, R.; Dieffenbach, A.K.; Lu, C.; Alves, A.C.; et al. Association of vitamin D status with arterial blood pressure and hypertension risk: A mendelian randomisation study. Lancet Diabetes Endocrinol. 2014, 2, 719–729. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.L.; Gu, H.B.; Zhang, Y.F.; Xia, Q.Q.; Qi, J.; Chen, J.C. Vitamin D supplementation in the treatment of chronic heart failure: A meta-analysis of randomized controlled trials. Clin. Cardiol. 2016, 39, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Mann, M.C.; Hobbs, A.J.; Hemmelgarn, B.R.; Roberts, D.J.; Ahmed, S.B.; Rabi, D.M. Effect of oral vitamin D analogs on mortality and cardiovascular outcomes among adults with chronic kidney disease: A meta-analysis. Clin. Kidney J. 2015, 8, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Yadav, A.K.; Singhal, M.; Kumar, V.; Lal, A.; Banerjee, D.; Gupta, K.L.; Jha, V. Vascular function and cholecalciferol supplementation in CKD: A self-controlled case series. J. Steroid Biochem. Mol. Biol. 2018, 180, 19–22. [Google Scholar] [CrossRef]
- Chitalia, N.; Ismail, T.; Tooth, L.; Boa, F.; Hampson, G.; Goldsmith, D.; Kaski, J.C.; Banerjee, D. Impact of vitamin D supplementation on arterial vasomotion, stiffness and endothelial biomarkers in chronic kidney disease patients. PLoS ONE 2014, 9, e91363. [Google Scholar] [CrossRef] [Green Version]
- Löfman, I.; Szummer, K.; Dahlström, U.; Jernberg, T.; Lund, L.H. Associations with and prognostic impact of chronic kidney disease in heart failure with preserved, mid-range, and reduced ejection fraction. Eur. J. Heart Fail. 2017, 19, 1606–1614. [Google Scholar] [CrossRef] [Green Version]
- Lundwall, K.; Jacobson, S.H.; Jörneskog, G.; Spaak, J. Treating endothelial dysfunction with vitamin D in chronic kidney disease: A metaanalysis. BMC Nephrol. 2018, 19, 247. [Google Scholar] [CrossRef]
- Chen, S.; Law, C.S.; Grigsby, C.L.; Olsen, K.; Hong, T.T.; Zhang, Y.; Yeghiazarians, Y.; Gardner, D.G. Cardiomyocyte-specific deletion of the vitamin D receptor gene results in cardiac hypertrophy. Circulation 2011, 124, 1838–1847. [Google Scholar] [CrossRef] [Green Version]
- Weishaar, R.E.; Simpson, R.U. Involvement of vitamin D3 with cardiovascular function. II. Direct and indirect effects. Am. J. Physiol. 1987, 253, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Weishaar, R.E.; Simpson, R.U. Vitamin D3 and cardiovascular function in rats. J. Clin. Investig. 1987, 79, 1706–1712. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Garami, M.; Cheng, T.; Gardner, D.G. 1.25 (OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J. Clin. Investig. 1996, 97, 1577–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, S.; Yalamarti, B.; Ke, Q.; Choudhury, S.; Yu, H.; Karumanchi, S.A.; Kroeger, P.; Thadhani, R.; Kang, P.M. Preventing progression of cardiac hypertrophy and development of heart failure by paricalcitol therapy in rats. Cardiovasc. Res. 2011, 91, 632–639. [Google Scholar] [CrossRef]
- Wang, A.Y.; Fang, F.; Chan, J.; Wen, Y.Y.; Qing, S.; Chan, I.H.; Lo, G.; Lai, K.N.; Lo, W.K.; Lam, C.W.; et al. Effect of paricalcitol on left ventricular mass and function in CKD—The OPERA trial. J. Am. Soc. Nephrol. 2014, 25, 175–186. [Google Scholar] [CrossRef]
- Thadhani, R.; Appelbaum, E.; Pritchett, Y.; Chang, Y.; Wenger, J.; Tamez, H.; Bhan, I.; Agarwal, R.; Zoccali, C.; Wanner, C.; et al. Vitamin D therapy and cardiac structure and function in patients with chronic kidney disease: The PRIMO randomized controlled trial. JAMA 2012, 307, 674–684. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.J.; Zhu, S.M.; Tang, F.L.; Zhu, X.S.; Fan, Z.D.; Wang, G.L.; Jiang, Y.F.; Zhang, Y. Effects of vitamin or its analogues on the mortality of patients with chronic kidney disease: An updated systematic review and meta-analysis. Eur. J. Clin. Nutr. 2017, 71, 683–693. [Google Scholar] [CrossRef]
- Shimada, T.; Yamazaki, Y.; Takahashi, M.; Hasegawa, H.; Urakawa, I.; Oshima, T.; Kakitani, M.; Tomizuka, K.; Fujita, T.; Fukumoto, S.; et al. Vitamin D receptor-intependent FGF23 actions in regulating phosphate and vitamin D metabolism. Am. J. Physiolol. Ren. Physiol. 2005, 289, 1088–1095. [Google Scholar] [CrossRef]
- Zhou, L.; Li, Y.; Zhou, D.; Tan, R.J.; Liu, Y. Loss of Klotho contributes to kidney injury by derepression of Wnt/β-catenin signaling. J. Am. Soc. Nephrol. 2013, 24, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, K.; Olauson, H.; Amin, R.; Ponnusamy, A.; Goetz, R.; Taylor, R.F.; Mohammadi, M.; Canfield, A.; Kublickiene, K.; Larsson, T.E. Arterial Klotho expression and FGF23 effects on vascular calcification and function. PLoS ONE 2013, 8, e60658. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Cha, S.K.; An, S.W.; Kuro, O.M.; Birnbaumer, L.; Huang, C.L. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat. Commun. 2012, 3, 1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isakova, T.; Wahl, P.; Vargas, G.S.; Gutierrez, O.-M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; Chen, J.; et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figurek, A.; Spasovski, G.; Popovic-Pejicic, S. FGF23 level and intima-media thickness are elevated from early stages of chronic kidney disease. Ther. Apher. Dial. 2018, 22, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Xu, J.; Zhang, S.; Jin, J. Meta-Analysis of the association between fibroblast growth factor 23 and mortality and cardiovascular events in hemodialysis patients. Blood Purif. 2019, 47, 24–30. [Google Scholar] [CrossRef]
- Isakova, T.; Cai, X.; Lee, J.; Xie, D.; Wang, X.; Mehta, R.; Allen, N.B.; Scialla, J.J.; Pencina, M.J.; Anderson, A.H.; et al. Longitudinal FGF23 trajectories and mortality in patients with CKD. J. Am. Soc. Nephrol. 2018, 29, 579–590. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.P.; Mendes, F.; Carias, E.; Goncalves, R.B.; Fragoso, A.; Dias, C.; Tavares, N.; Cafe, H.M.; Santos, N.; Rato, F.; et al. Plasmatic Klotho and FGF23 levels as biomarkers of CKD-associated cardiac disease in type 2 diabetic patients. Int. J. Mol. Sci. 2019, 20, 1536. [Google Scholar] [CrossRef] [Green Version]
- Dai, B.; David, V.; Martin, A.; Huang, J.; Li, H.; Jiao, Y.; Gu, W.; Quarles, L.D. A comparative transcriptome analysis identifying FGF23 regulated genes in the kidney of a mouse CKD model. PLoS ONE 2012, 7, e44161. [Google Scholar] [CrossRef] [Green Version]
- Leifheit-Nestler, M.; Kirchhoff, F.; Nespor, J.; Richter, B.; Soetje, B.; Klintschar, M.; Heineke, J.; Haffner, D. Fibroblast growth factor 23 is induced by an activated renin-angiotensin-aldosterone system in cardiac myocytes and promotes the pro-fibrotic crosstalk between cardiac myocytes and fibroblasts. Nephrol. Dial. Transpl. 2018, 33, 1722–1734. [Google Scholar] [CrossRef]
- Agoro, R.; Montagna, A.; Goetz, R.; Aligbe, O.; Singh, G.; Coe, L.M.; Mohammadi, M.; Rivella, S.; Sitara, D. Inhibition of fibroblast growth factor 23 (FGF23) signaling rescues renal anemia. FASEB J. 2018, 32, 3752–3764. [Google Scholar] [CrossRef] [Green Version]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutierrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Cai, C.; Xiao, Z.; Quarles, L.D. FGF23 induced left ventricular hypertrophy mediated by FGFR4 signaling in the myocardium is attenuated by soluble Klotho in mice. J. Mol. Cell Cardiol. 2019, 21, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Matsui, I.; Oka, T.; Kusunoki, Y.; Mori, D.; Hashimoto, N.; Matsumoto, A.; Shimada, K.; Yamaguchi, S.; Kubota, K.; Yonemoto, S.; et al. Cardiac hypertrophy elevates serum levels of fibroblast growth factor 23. Kidney Int. 2018, 94, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Yeung, S.M.H.; Binnenmars, S.H.; Gant, C.M.; Navis, G.; Gansevoort, R.T.; Bakker, S.J.L.; De Brost, M.H.; Laverman, G.D. Fibroblast growth factor 23 and mortality in patients with type 2 diabetes and normal or mildly impaired kidney function. Diabetes Care 2019, 42, 2151–2153. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.L.; Plesner, L.L.; Warming, P.E.; Mortensen, O.H.; Iversen, K.K.; Heaf, J.G. FGF23 in hemodialysis patients is associated with left ventricular hypertrophy and reduced ejection fraction. Nefrologia 2019, 39, 258–268. [Google Scholar] [CrossRef]
- Gruson, D.; Ferracin, B.; Ahn, S.S.; Rousseau, M.F. Comparison of fibroblast growth factor 23, soluble ST2 and Galectin-3 for prognostication of cardiovascular death in heart failure patients. Int. J. Cardiol. 2015, 189, 185–187. [Google Scholar] [CrossRef]
- Grabner, A.; Schramm, K.; Silswal, N.; Hendrix, M.; Yanucil, C.; Czaya, B.; Singh, S.; Wolf, M.; Hermann, S.; Stypmann, J.; et al. FGF23/FGFR4-mediated left ventricular hypertrophy is reversible. Sci. Rep. 2017, 16, 1993. [Google Scholar] [CrossRef]
- Leifheit-Nestler, M.; Grabner, A.; Hermann, L.; Richter, B.; Schmitz, K.; Fischer, D.C.; Yanucil, C.; Faul, C.; Haffner, D. Vitamin D treatment attenuates cardiac FGF23/FGFR4 signaling and hypertrophy in uremic rats. Nephrol. Dial. Transpl. 2017, 32, 1493–1503. [Google Scholar] [CrossRef]
- Francis, C.; Courbon, G.; Gerber, C.; Neuburg, S.; Wang, X.; Dussold, C.; Capella, M.; Qi, L.; Isakova, T.; Mehta, R.; et al. Ferric citrate reduces fibroblast growth factor 23 levels and improves renal and cardiac function in a mouse model of chronic kidney disease. Kidney Int. 2019, 96, 1346–1358. [Google Scholar] [CrossRef]
- Neyra, J.A.; Hu, M.C. Potential application of klotho in human chronic kidney disease. Bone 2017, 100, 41–49. [Google Scholar] [CrossRef]
- Kovesdy, C.P.; Quarles, L.D. The role of fibroblast growth factor-23 in cardiorenal syndrome. Nephron Clin. Pract. 2013, 123, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Yoon, J.; An, S.W.; Kuro-o, M.; Huang, C.L. Soluble Klotho Protects against Uremic Cardiomyopathy Independently of Fibroblast Growth Factor 23 and Phosphate. J. Am. Soc. Nephrol. 2015, 26, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhu, L.J.; Waaga-Gasser, A.M.; Ding, Y.; Cao, M.; Jadhav, S.J.; Kirollos, S.; Shekar, P.S.; Padera, R.F.; Chang, Y.C.; et al. The axis of local cardiac endogenous Klotho-TGF-β1-Wnt signaling mediates cardiac fibrosis in human. J. Mol. Cell Cardiol. 2019, 136, 113–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, M.E.; De Las Fuentes, L.; Ginsberg, C.; Ginsberg, C.; Rothstein, M.; Dietzen, D.J.; Cheng, S.C.; Ross, W.; Windus, D.; Davila-Roman, V.G.; et al. Left ventricular mass progression despite stable blood pressure and kidney function in stage 3 chronic kidney disease. Am. J. Nephrol. 2014, 39, 392–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memmos, E.; Sarafidis, P.; Pateinakis, P.; Tsiantoulas, A.; Faitatzidou, D.; Giamalis, P.; Vasilikos, V.; Papagianni, A. Soluble Klotho is associated with mortality and cardiovascular events in hemodialysis. BMC Nephrol. 2019, 11, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandenburg, V.M.; Kleber, M.E.; Vervloet, M.G.; Larsson, T.E.; Tomaschitz, A.; Pilz, S.; Stojakovic, T.; Delgado, G.; Grammer, T.B.; Marx, N.; et al. Soluble klotho and mortality: The Ludwigshafen Risk and Cardiovascular Health Study. Atherosclerosis 2015, 242, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kang, E.; Oh, Y.K.; Kim, Y.H.; Han, S.H.; Yoo, T.H.; Chae, D.W.; Lee, J.; Ahn, C.; Oh, K.H.; et al. The association between soluble klotho and cardiovascular parameters in chronic kidney disease: Results from the KNOW-CKD study. BMC Nephrol. 2018, 5, 51. [Google Scholar] [CrossRef]
- Smith, E.R.; Holt, S.G.; Hewitson, T.D. αKlotho-FGF23 interactions and their role in kidney disease: A molecular insight. Cell Mol. Life Sci. 2019, 76, 4705–4724. [Google Scholar] [CrossRef]
- Li, F.; Yao, Q.; Ao, L.; Cleveland, J.C., Jr.; Dong, N.; Fullerton, D.A.; Meng, X. Klotho suppresses high phosphate-induced osteogenic responses in human aortic valve interstitial cells through inhibition of Sox9. J. Mol. Med. 2017, 95, 739–751. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Gillings, N.; Flores, B.; Takahashi, M.; Kuro-O, M.; Moe, O.W. Recombinant α-Klotho may be prophylactic and therapeutic for acute to chronic kidney disease progression and uremic cardiomyopathy. Kidney Int. 2017, 91, 1104–1114. [Google Scholar] [CrossRef] [Green Version]
- Seiler, S.; Rogacev, K.S.; Roth, H.J.; Shafein, P.; Emrich, I.; Neuhaus, S.; Floege, J.; Fliser, D.; Heine, G.H. Associations of FGF-23 and sKlotho with cardiovascular outcomes among patients with CKD stages 2-4. Clin. J. Am. Soc. Nephrol. 2014, 6, 1049–1058. [Google Scholar] [CrossRef]
- Lu, X.; Hu, M.C. Klotho/FGF23 Axis in Chronic Kidney Disease and Cardiovascular Disease. Kidney Dis. 2017, 3, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, D.G.; Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Hayes, T.; Skonier, J.E.; Shpektor, D.; Jonas, M.; Kovacevich, B.R.; Staehling-Hampton, K.; et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003, 22, 6267–6276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.X.; Baum, W.; Dwyer, D.; Stock, M.; Schwabe, K.; Ke, H.Z.; Stolina, M.; Schett, G.; Bozec, A. Sclerostin inhibition reverses systemic periarticular and local bone loss in arthritis. Ann. Rheum. Dis. 2013, 72, 1732–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Bezooijen, R.L.; Ten Dijke, P.; Papapoulos, S.E.; Lowik, C.W. SOST/sclerostin, an osteocyte-derived negative regulator of bone formation. Cytokine Growth Factor Rev. 2005, 6, 319–327. [Google Scholar] [CrossRef]
- Van Bezooijen, R.L.; Bronckers, A.L.; Gortzak, R.A.; Hogendoorn, P.C.W.; Van der Wee-Pals, L.; Balemans, W.; Oostenbroek, H.J.; Van Hul, W.; Hamersma, H.; Dikkers, F.G.; et al. Sclerostin in mineralised matrices and van Buchem disease. J. Dent. Res. 2009, 88, 569–574. [Google Scholar] [CrossRef]
- Winkler, D.G.; Sutherland, M.S.; Ojala, E.; Turcott, E.; Geoghegan, J.C.; Shpektor, D.; Skonier, J.E.; Yu, C.; Latham, J.A. Sclerostin inhibition of Wnt-3a-induced C3H10T1/2 cell differentiation is indirect and mediated by bone morphogenetic proteins. J. Biol. Chem. 2005, 280, 2498–2502. [Google Scholar] [CrossRef] [Green Version]
- Brunkow, M.E.; Gardner, J.C.; Van Ness, J.; Paeper, B.W.; Kovacevich, B.R.; Proll, S.; Skonier, J.E.; Zhao, L.; Sabo, P.J.; Fu, Y.; et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot containing protein. Am. J. Hum. Genet. 2001, 68, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Balemans, W.; Ebeling, M.; Patel, N.; Van Hul, E.; Olson, P.; Dioszegi, M.; Lacza, C.; Wuyts, W.; Van Den Ende, J.; Willems, P.; et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum. Mol. Genet. 2001, 10, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Anderson, L.; Seilhamer, J. A comparison of selected mRNA and protein abundances in human liver. Electrophoresis 1997, 18, 533–537. [Google Scholar] [CrossRef]
- Figurek, A.; Spasovski, G. Is serum sclerostin a marker of atherosclerosis in patients with chronic kidney disease-mineral and bone disorder? Int. Urol. Nephol. 2018, 50, 1863–1870. [Google Scholar] [CrossRef]
- Koos, R.; Brandenburg, V.; Mahnken, A.H.; Schneider, R.; Dohmen, G.; Autschbach, R.; Marx, N.; Kramann, R. Sclerostin as a potential novel biomarker for aortic valve calcification: An in-vivo and ex-vivo study. J. Heart Valve Dis. 2013, 22, 317–325. [Google Scholar] [PubMed]
- Ji, Y.Q.; Guan, L.N.; Yu, S.X.; Yin, P.Y.; Shen, X.Q.; Sun, Z.W.; Liu, J.; Lv, W.; Yu, G.P.; Ren, C.; et al. Serum sclerostin as a potential novel biomarker for heart valve calcification in patients with chronic kidney disease. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8822–8829. [Google Scholar] [PubMed]
- Desjardins, L.; Liabeuf, S.; Oliveira, R.B.; Louvet, L.; Kamel, S.; Lemke, H.D.; Vanholder, R.; Choukroun, G.; Massy, Z.A.; European Uremic Toxin (EuTox) Work Group. Uremic toxicity and sclerostin in chronic kidney disease patients. Nephrol. Ther. 2014, 10, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Stavrinou, E.; Sarafidis, P.A.; Koumaras, C.; Loutradis, C.; Giamalis, P.; Tziomalos, K.; Karagiannis, A.; Papagianni, A. Increased Sclerostin, but Not Dickkopf-1 Protein, Is Associated with Elevated Pulse Wave Velocity in Hemodialysis Subjects. Kidney Blood Press. Res. 2019, 44, 679–689. [Google Scholar] [CrossRef]
- Kirkpantur, A.; Balci, M.; Turkvatan, A.; Afsar, B. Serum sclerostin levels, arteriovenous fistula calcification and 2-years all-cause mortality in prevalent hemodialysis patients. Nefrologia 2016, 36, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Kalousova, M.; Dusilova-Sulkova, S.; Kubena, A.A.; Zakiyanov, O.; Tesar, V.; Zima, T. Sclerostin levels predict cardiovascular mortality in long-term hemodialysis patients: A prospective observational cohort study. Physiol. Res. 2019, 29, 547–558. [Google Scholar] [CrossRef]
- Liao, R.; Wang, L.; Li, J.; Sun, S.; Xiong, Y.; Li, Y.; Han, M.; Jiang, H.; Anil, M.; Su, B.; et al. Vascular calcification is associated with Wnt-signaling pathway and blood pressure variability in chronic kidney disease rats. Nephrology 2019. [Google Scholar] [CrossRef]
- Jorgensen, H.S.; Winther, S.; Dupont, L.; Bottcher, M.; Rejnmark, L.; Hauge, E.M.; Svensson, M.; Ivarsen, P. Sclerostin is not associated with cardiovascular event or fracture in kidney transplantation candidates. Clin. Nephrol. 2018, 90, 18–26. [Google Scholar] [CrossRef]
- Kanbay, M.; Solak, Y.; Siriopol, D.; Aslan, G.; Afsar, B.; Yazici, D.; Covic, A. Sclerostin, cardiovascular disease and mortality: A systematic review and meta-analysis. Int. Urol. Nephrol. 2016, 48, 2029–2042. [Google Scholar] [CrossRef]
- Kanbay, M.; Siriopol, D.; Saglam, M.; Kurt, Y.G.; Gok, M.; Cetinkay, H.; Karaman, M.; Unal, H.U.; Oguz, Y.; Sari, S.; et al. Serum sclerostin and adverse outcomes in nondialyzed chronic kidney disease patients. J. Clin. Endocrinol. Metab. 2014, 99, E1854–E1861. [Google Scholar] [CrossRef] [Green Version]
- Drechsler, C.; Evenepoel, P.; Vervloet, M.G.; Wanner, C.; Ketteler, M.; Marx, N.; Floege, J.; Dekker, F.W.; Brandenburg, V.M. NECOSAD Study Group. High levels of circulating sclerostin are associated with better cardiovascular survival in incident dialysis patients: Results from the NECOSAD study. Nephrol. Dial. Transpl. 2015, 30, 288–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClung, M.R. Sclerostin antibodies in osteoporosis: Latest evidence and therapeutic potential. Ther. Adv. Musculoskelet Dis. 2017, 9, 263–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, F.; Cai, X.; Yang, W.; Gao, L.; Chen, L.; Wu, J.; Lingong, J. Denosumab or romosozumab therapy and risk of cardiovascular events in patients with primary osteoporosis: Systematic review and meta-analysis. Bone 2020, 130, 115121. [Google Scholar] [CrossRef] [PubMed]
- Persy, V.; D’Haese, P. Vascular calcification and bone disease: The calcification paradox. Trends Mol. Med. 2009, 15, 405–406. [Google Scholar] [CrossRef]
- Collin-Osdoby, P. Regulation of vascular calcification by osteoclast regulatory factors RANKL and osteoprotegerin. Circ. Res. 2004, 95, 1046–1057. [Google Scholar] [CrossRef]
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L.; et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes. Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef]
- Min, H.; Morony, S.; Sarosi, I.; Dunstan, C.R.; Capparelli, C.; Scully, S.; Van, G.; Kaufman, S.; Kostenuik, P.J.; Lacey, D.L.; et al. Osteoprotegerin reverses osteoporosis by inhibiting endosteal osteoclasts and prevents vascular calcification by blocking a process resembling osteoclastogenesis. J. Exp. Med. 2000, 192, 463–474. [Google Scholar] [CrossRef] [Green Version]
- Tyson, K.L.; Reynolds, J.L.; McNair, R.; Zhang, Q.; Weissberg, P.L.; Shanahan, C.M. Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 489–494. [Google Scholar] [CrossRef] [Green Version]
- Ozkok, A.; Caliskan, Y.; Sakaci, T.; Erten, G.; Karahan, G.; Ozel, A.; Unsal, A.; Yildiz, A. Osteoprotegerin/RANKL axis and progression of coronary artery calcification in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2012, 7, 965–973. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| FGF-23–Klotho–Sclerostin Axis | Cellular Level | Tissue Level | Circulation | Clinical Observation | Therapeutic Potential |
|---|---|---|---|---|---|
| FGF-23 | FGF23 directly induces LVH by binding to the FGFR-4 in cardiomyocytes RAAS activation induces FGF23 expression in cardiac myocytes and stimulates pro-fibrotic crosstalk between cardiac myocytes and fibroblasts | FGF23 increases production of TGF-β, lipocalin-2, and TNF-α, and thus promoting the inflammation process | LVH is shown to be associated with an increase in both myocardial and serum intact FGF23 FGF23 contributes to renal anemia development -> contribution to LVH aggravation | FGF23 levels correlate positively with LVH and negatively to left ventricular ejection fraction in patients on hemodialysis | Vitamin D treatment reduces LVH Ferric citrate lowers FGF23 levels and improves cardiac function and patient survival |
| Klotho | Cardioprotective effect by downregulation of TRPC6 channels in cardiomyocytes, important for angiotensin II-induced hypertrophy signaling Klotho upregulation inhibits TGF-β1-induced fibrosis and pathogenic Wnt/ β-catenin signaling in cardiomyocytes | Cardiomyocytes and cardiac fibroblasts express klotho | Uremic serum or TGF-β1 suppressed klotho expression by cardiomyocytes | FGF23/klotho ratio correlates with changes in left ventricular mass Low klotho levels are associated with CV events Serum klotho is an independent biomarker of a left ventricular mass index | Klotho administration attenuates high-phosphate induced renal and cardiac fibrosis and improved both renal and cardiac function |
| Sclerostin | Lacking data about the association with LVH | Lacking data | Lacking data | Elevated serum sclerostin levels in patients with aortic valve calcification with increased upregulation of sclerostin mRNA | Not yet clear whether therapeutic decrease of sclerostin levels is beneficial or deleterious for CV outcome |
| CKD-MBD Biomarkers | Role in Bone Metabolism | Vascular Calcification | Uremic Cardiomyopathy |
|---|---|---|---|
| Phosphate | Major trigger in CKD-MBD ↑P →↑PTH→ ↑Vit D →↑Ca ↑P →↑FGF23→↓Vit D→↓ Ca | Promotes VC Impairs endothelial function | Cardiac fibrosis |
| PTH | Key mediator of bone turnover Regulates P and Ca homeostasis | Complex paracrine and systemic effectPromotes VC Impairs endothelial function | Cardiac electrophysiology Cardiomyocyte hypertrophy Cardiac interstitial fibrosis |
| Vit D | Key role in Ca, P homeostasis Depletion promote sHPTH and osteitis fibrosis cystica | Biphasic curve of Vit D on calcification | Increases collagen ↓Vit D→ impairs contractile function Increases cardiac mass |
| Klotho | Acts as a Wnt-inhibitor Modify bone metabolism | Inhibitor of VC Klotho deficiency→ impair endothelial function | Klotho deficiency→ LVH Cardiac fibrosis |
| FGF23 | Posphaturic hormone acts through α-klotho | Is not clear if it has a direct effect on VC | Concentric hypertrophy |
| Sclerostin | Inhibits bone turnover | Marker of vascular calcification | There are no conclusive data |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rroji, M.; Figurek, A.; Spasovski, G. Should We Consider the Cardiovascular System While Evaluating CKD-MBD? Toxins 2020, 12, 140. https://doi.org/10.3390/toxins12030140
Rroji M, Figurek A, Spasovski G. Should We Consider the Cardiovascular System While Evaluating CKD-MBD? Toxins. 2020; 12(3):140. https://doi.org/10.3390/toxins12030140
Chicago/Turabian StyleRroji, Merita, Andreja Figurek, and Goce Spasovski. 2020. "Should We Consider the Cardiovascular System While Evaluating CKD-MBD?" Toxins 12, no. 3: 140. https://doi.org/10.3390/toxins12030140
APA StyleRroji, M., Figurek, A., & Spasovski, G. (2020). Should We Consider the Cardiovascular System While Evaluating CKD-MBD? Toxins, 12(3), 140. https://doi.org/10.3390/toxins12030140