Cardiovascular Calcification in Chronic Kidney Disease—Therapeutic Opportunities


Abstract
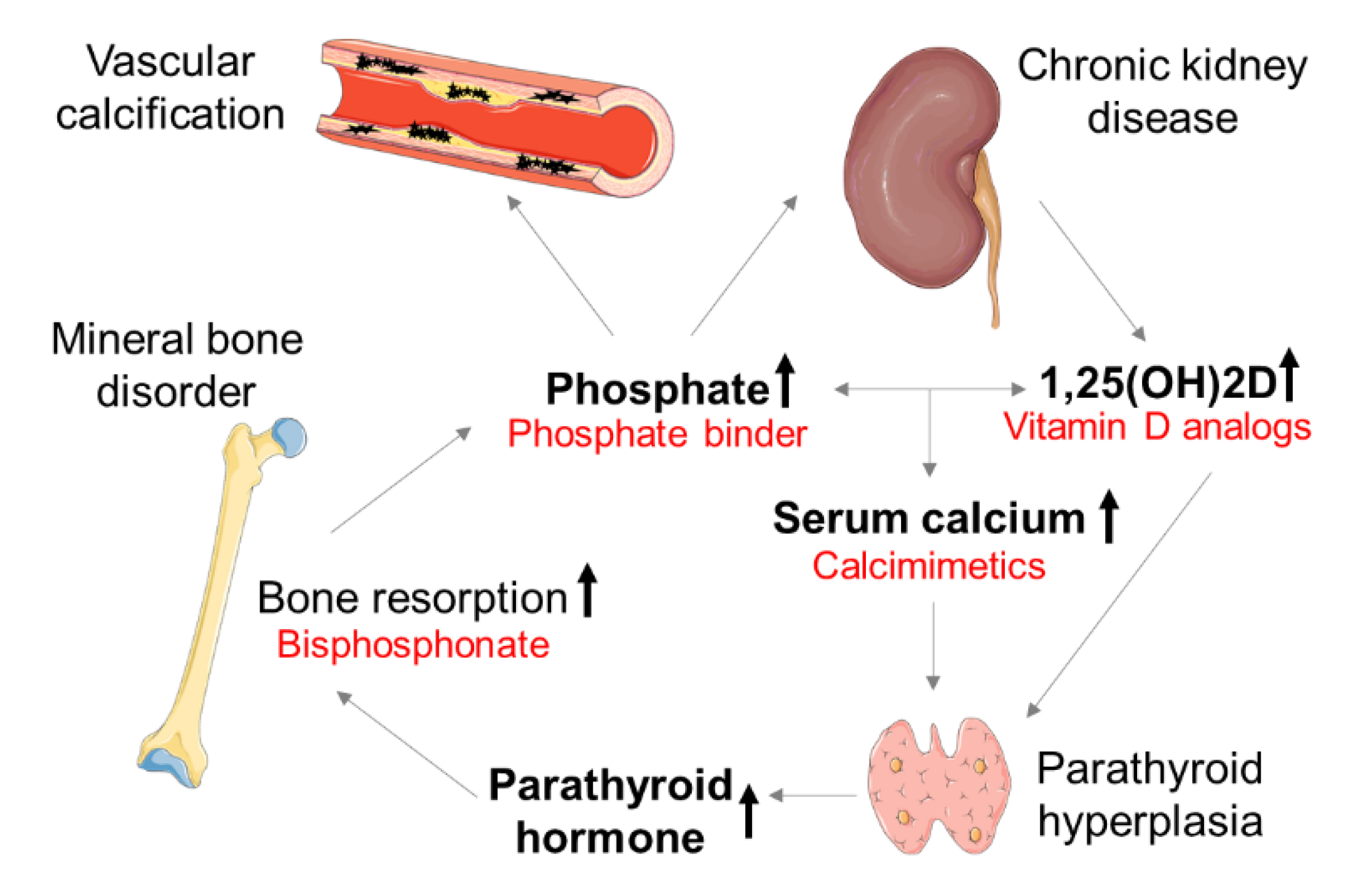
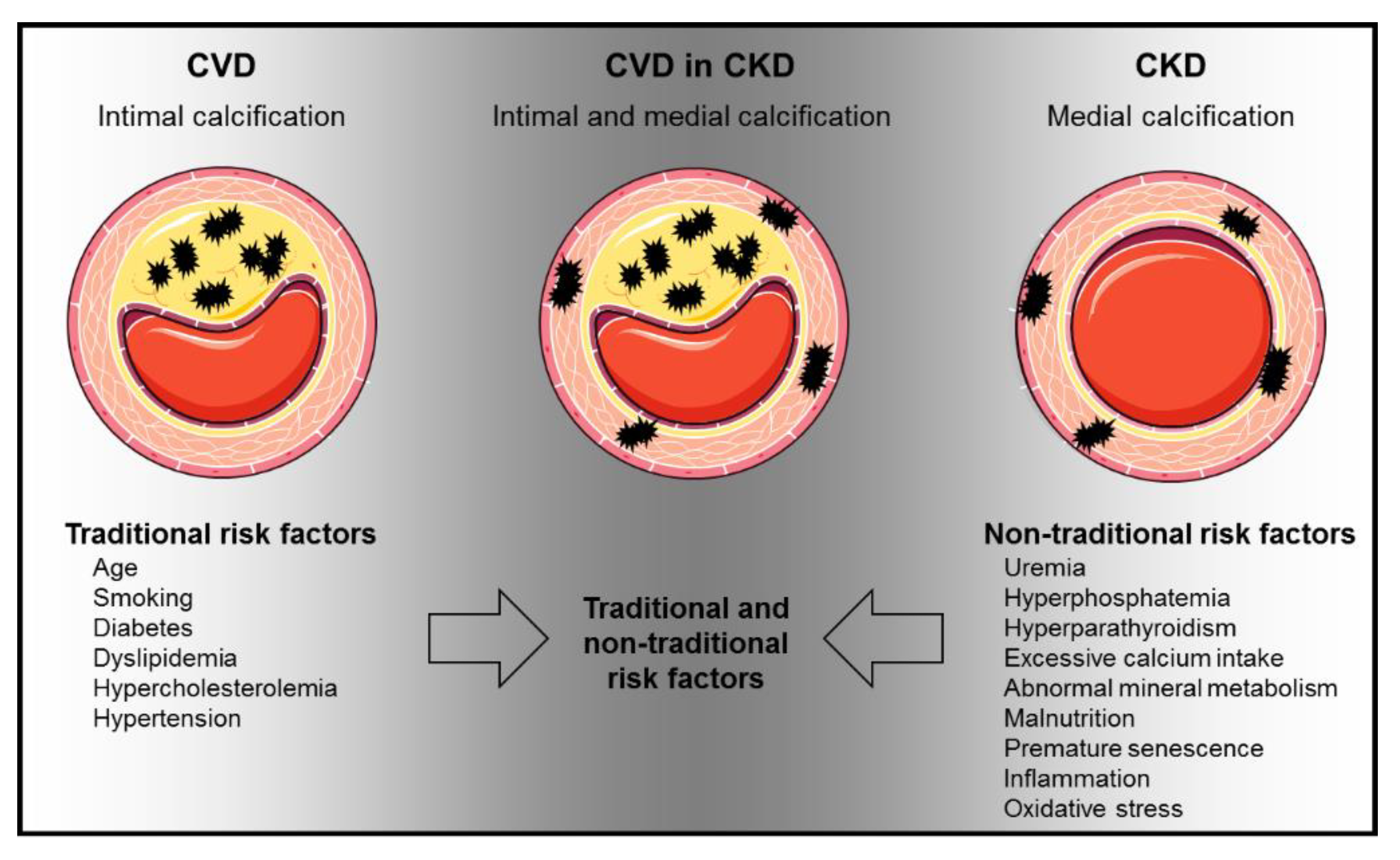
:1. Introduction
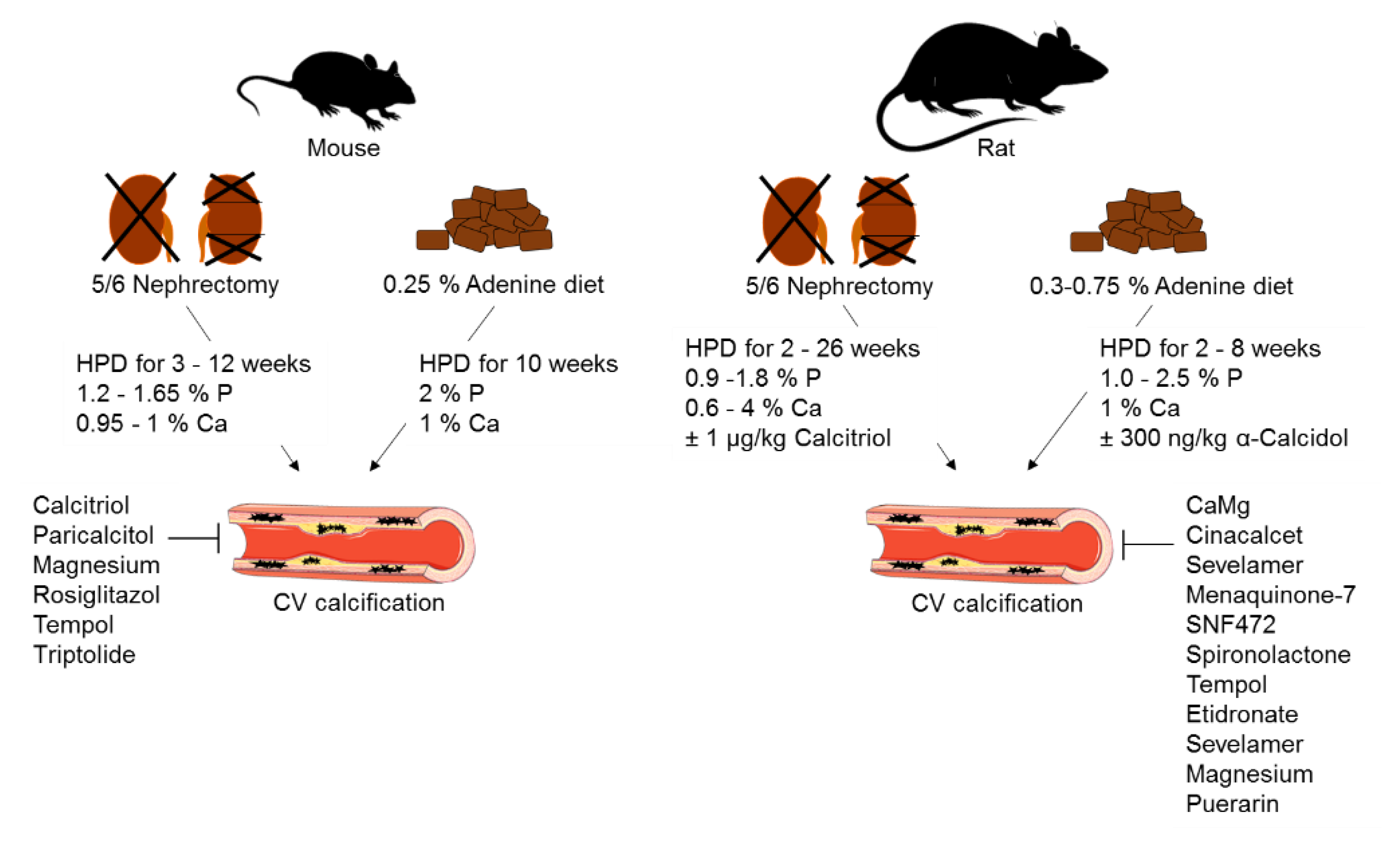
2. Animal Models of CKD
3. Therapeutic Concepts of CV Calcification in CKD
3.1. Phosphate Binder
3.2. Calcimimetics
4. Novel Therapeutic Strategies—from Experimental Models to the Clinic
4.1. Bisphosphonates

{kind=link}
{kind=link}
{kind=link}
| Treatment | Substance | Dosis | Application | Experimental Model | Species, Strain | Ref. |
|---|---|---|---|---|---|---|
| Bisphospho-nate | Etidronate | 5 or 10 mg/kg | s.c., daily, 3 weeks | 5/6 nephrectomy | Wistar rat | [54] |
| Vitamin K | Mena-quinone-7 | 50 µg/kg | Oral gavage, daily 4 weeks | Adenine diet | Sprague-Dawley rat | [66] |
| Omega-3 fatty acid | Eicosapenta-enoic acid | 300 mg/kg | Oral gavage, daily 4 weeks | Adenine diet | Sprague-Dawley rat | [66] |
| Vitamin D receptor agonist | Calcitriol Paricalcitol | 30 ng/kg 100 or 300 ng/kg | i.p., 3 times/week, 3 weeks | 5/6 nephrectomy | DBA/2J mouse | [26] |
| Dietary supplement | Magnesium | 0.1–1.1% | Food intake, 14 days | 5/6 nephrectomy | Wistar rat | [67] |
| Dietary supplement | Magnesium | 3% | Food intake, 7 weeks | 5/6 nephrectomy | Non-agouti mouse | [68] |
| Hexasodium salt | SNF472 | 50 mg/kg | i.v., daily, 19 days | Adenine diet | Wistar rat | [20] |
4.2. Vitamin K
4.3. Vitamin D
4.4. Magnesium
4.5. Hexasodium Salt of Myo-Inositol Hexaphosphate
5. Promising Treatments of CV Calcification in Experimental CKD Models
| Treatment | Substance | Dosis | Application | Experimental Model | Species, Strain | Ref. |
|---|---|---|---|---|---|---|
| Isoflavonoid | Puerarin | 400 mg/kg | Oral gavage, daily; 4 weeks | 5/6 nephrectomy | Sprague-Dawley rat | [104] |
| PPARγ agonist | Rosiglitazol | 10 mg/kg | Oral gavage, daily; 12 weeks | 5/6 nephrectomy | DBA/2J mouse | [107] |
| NF-κB inhibitor | Tempol | 3 mmol/L | Drinking water; 10 weeks | Adenine diet | DBA/2J mouse | [109] |
| NF-κB inhibitor | Tempol | 3 mmol/L | Drinking water; 6 weeks | Adenine diet | Sprague-Dawley rat | [110] |
| NF-κB inhibitor | Triptolide | 70 µg/kg | i.p., daily; 10 weeks | Adenine diet | DBA/2J mouse | [109] |
| MR antagonist | Spirono-lactone | 100 mg/kg | Food intake, daily; 2 weeks | Adenine diet | Sprague-Dawley rat | [111] |
6. Potential Diagnostic Tools for CV Calcification in CKD
6.1. Development of the T50 Assay
6.2. Clinical Association
7. Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Metra, M.; Zaca, V.; Parati, G.; Agostoni, P.; Bonadies, M.; Ciccone, M.; Cas, A.D.; Iacoviello, M.; Lagioia, R.; Lombardi, C.; et al. Cardiovascular and noncardiovascular comorbidities in patients with chronic heart failure. J. Cardiovasc. Med. 2011, 12, 76–84. [Google Scholar] [CrossRef]
- Noels, H.; Boor, P.; Goettsch, C.; Hohl, M.; Jahnen-Dechent, W.; Jankowski, V.; Kindermann, I.; Kramann, R.; Lehrke, M.; Linz, D.; et al. The new SFB/TRR219 Research Centre. Eur. Heart J. 2018, 39, 975–977. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Yoshida, M. Protein-bound uremic toxins: New culprits of cardiovascular events in chronic kidney disease patients. Toxins 2014, 6, 665–678. [Google Scholar] [CrossRef] [Green Version]
- Lekawanvijit, S.; Kompa, A.R.; Wang, B.H.; Kelly, D.J.; Krum, H. Cardiorenal syndrome: The emerging role of protein-bound uremic toxins. Circ. Res. 2012, 111, 1470–1483. [Google Scholar] [CrossRef] [Green Version]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [Green Version]
- Schlieper, G.; Schurgers, L.; Brandenburg, V.; Reutelingsperger, C.; Floege, J. Vascular calcification in chronic kidney disease: An update. Nephrol. Dial. Transplant. 2016, 31, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Viegas, C.; Araujo, N.; Marreiros, C.; Simes, D. The interplay between mineral metabolism, vascular calcification and inflammation in Chronic Kidney Disease (CKD): Challenging old concepts with new facts. Aging (Albany NY) 2019, 11, 4274–4299. [Google Scholar] [CrossRef]
- Dayanand, P.; Sandhyavenu, H.; Dayanand, S.; Martinez, J.; Rangaswami, J. Role of Bisphosphonates in Vascular calcification and Bone Metabolism: A Clinical Summary. Curr. Cardiol. Rev. 2018, 14, 192–199. [Google Scholar] [CrossRef]
- Kiel, D.P.; Kauppila, L.I.; Cupples, L.A.; Hannan, M.T.; O’Donnell, C.J.; Wilson, P.W. Bone loss and the progression of abdominal aortic calcification over a 25 year period: The Framingham Heart Study. Calcif. Tissue Int. 2001, 68, 271–276. [Google Scholar] [CrossRef]
- Tanko, L.B.; Christiansen, C.; Cox, D.A.; Geiger, M.J.; McNabb, M.A.; Cummings, S.R. Relationship between osteoporosis and cardiovascular disease in postmenopausal women. J. Bone Miner. Res. 2005, 20, 1912–1920. [Google Scholar] [CrossRef] [Green Version]
- Beto, J.; Bhatt, N.; Gerbeling, T.; Patel, C.; Drayer, D. Overview of the 2017 KDIGO CKD-MBD Update: Practice Implications for Adult Hemodialysis Patients. J. Ren. Nutr. 2019, 29, 2–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, N.; Noels, H.; Jankowski, J.; Floege, J.; Fliser, D.; Bohm, M. Mechanisms of cardiovascular complications in chronic kidney disease: Research focus of the Transregional Research Consortium SFB TRR219 of the University Hospital Aachen (RWTH) and the Saarland University. Clin. Res. Cardiol. 2018, 107, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.S.; Blaha, M.J.; Blankstein, R.; Agatston, A.; Rivera, J.J.; Virani, S.S.; Ouyang, P.; Jones, S.R.; Blumenthal, R.S.; Budoff, M.J.; et al. Dyslipidemia, coronary artery calcium, and incident atherosclerotic cardiovascular disease: Implications for statin therapy from the multi-ethnic study of atherosclerosis. Circulation 2014, 129, 77–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutcheson, J.D.; Goettsch, C.; Bertazzo, S.; Maldonado, N.; Ruiz, J.L.; Goh, W.; Yabusaki, K.; Faits, T.; Bouten, C.; Franck, G.; et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat. Mater. 2016, 15, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Shroff, R.C.; Shanahan, C.M. The vascular biology of calcification. Semin. Dial. 2007, 20, 103–109. [Google Scholar] [CrossRef]
- Schlieper, G.; Hess, K.; Floege, J.; Marx, N. The vulnerable patient with chronic kidney disease. Nephrol. Dial. Transplant. 2016, 31, 382–390. [Google Scholar] [CrossRef] [Green Version]
- Ketteler, M.; Schlieper, G.; Floege, J. Calcification and cardiovascular health: New insights into an old phenomenon. Hypertension 2006, 47, 1027–1034. [Google Scholar] [CrossRef]
- Shroff, R.; Long, D.A.; Shanahan, C. Mechanistic insights into vascular calcification in CKD. J. Am. Soc. Nephrol. 2013, 24, 179–189. [Google Scholar] [CrossRef]
- Nitta, K.; Ogawa, T.; Hanafusa, N.; Tsuchiya, K. Recent Advances in the Management of Vascular Calcification in Patients with End-Stage Renal Disease. Contrib. Nephrol. 2019, 198, 62–72. [Google Scholar] [CrossRef]
- Ferrer, M.D.; Ketteler, M.; Tur, F.; Tur, E.; Isern, B.; Salcedo, C.; Joubert, P.H.; Behets, G.J.; Neven, E.; D’Haese, P.C.; et al. Characterization of SNF472 pharmacokinetics and efficacy in uremic and non-uremic rats models of cardiovascular calcification. PLoS ONE 2018, 13, e0197061. [Google Scholar] [CrossRef] [Green Version]
- Raggi, P.; Bellasi, A.; Bushinsky, D.; Bover, J.; Rodriguez, M.; Ketteler, M.; Sinha, S.; Salcedo, C.; Gillotti, K.; Padgett, C.; et al. Slowing Progression of Cardiovascular Calcification with SNF472 in Patients on Hemodialysis: Results of a Randomized, Phase 2b Study. Circulation 2019. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, T.D.; Holt, S.G.; Smith, E.R. Animal models to study links between cardiovascular disease and renal failure and their relevance to human pathology. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, G.J.; Hewitson, T.D. Animal models of chronic kidney disease: Useful but not perfect. Nephrol. Dial. Transplant. 2013, 28, 2432–2438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shobeiri, N.; Adams, M.A.; Holden, R.M. Vascular calcification in animal models of CKD: A review. Am. J. Nephrol. 2010, 31, 471–481. [Google Scholar] [CrossRef]
- Yokozawa, T.; Oura, H.; Okada, T. Metabolic effects of dietary purine in rats. J. Nutr. Sci. Vitaminol. 1982, 28, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Lau, W.L.; Leaf, E.M.; Hu, M.C.; Takeno, M.M.; Kuro-o, M.; Moe, O.W.; Giachelli, C.M. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. 2012, 82, 1261–1270. [Google Scholar] [CrossRef] [Green Version]
- Rabe, M.; Schaefer, F. Non-Transgenic Mouse Models of Kidney Disease. Nephron 2016, 133, 53–61. [Google Scholar] [CrossRef]
- El-Abbadi, M.M.; Pai, A.S.; Leaf, E.M.; Yang, H.Y.; Bartley, B.A.; Quan, K.K.; Ingalls, C.M.; Liao, H.W.; Giachelli, C.M. Phosphate feeding induces arterial medial calcification in uremic mice: Role of serum phosphorus, fibroblast growth factor-23, and osteopontin. Kidney Int. 2009, 75, 1297–1307. [Google Scholar] [CrossRef] [Green Version]
- Makaryus, A.N.; Sison, C.; Kohansieh, M.; Makaryus, J.N. Implications of gender difference in coronary calcification as assessed by ct coronary angiography. Clin. Med. Insights Cardiol. 2014, 2014, 51–55. [Google Scholar] [CrossRef]
- Ishola, D.A., Jr.; van der Giezen, D.M.; Hahnel, B.; Goldschmeding, R.; Kriz, W.; Koomans, H.A.; Joles, J.A. In mice, proteinuria and renal inflammatory responses to albumin overload are strain-dependent. Nephrol. Dial. Transplant. 2006, 21, 591–597. [Google Scholar] [CrossRef] [Green Version]
- Cozzolino, M.; Staniforth, M.E.; Liapis, H.; Finch, J.; Burke, S.K.; Dusso, A.S.; Slatopolsky, E. Sevelamer hydrochloride attenuates kidney and cardiovascular calcifications in long-term experimental uremia. Kidney Int. 2003, 64, 1653–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Schutter, T.M.; Behets, G.J.; Geryl, H.; Peter, M.E.; Steppan, S.; Gundlach, K.; Passlick-Deetjen, J.; D’Haese, P.C.; Neven, E. Effect of a magnesium-based phosphate binder on medial calcification in a rat model of uremia. Kidney Int. 2013, 83, 1109–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neven, E.; De Schutter, T.M.; Dams, G.; Gundlach, K.; Steppan, S.; Buchel, J.; Passlick-Deetjen, J.; D’Haese, P.C.; Behets, G.J. A magnesium based phosphate binder reduces vascular calcification without affecting bone in chronic renal failure rats. PLoS ONE 2014, 9, e107067. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.C.; Gardner, S.; Tonelli, M.; Mavridis, D.; Johnson, D.W.; Craig, J.C.; French, R.; Ruospo, M.; Strippoli, G.F. Phosphate-Binding Agents in Adults With CKD: A Network Meta-analysis of Randomized Trials. Am. J. Kidney Dis. 2016, 68, 691–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamal, S.A.; Vandermeer, B.; Raggi, P.; Mendelssohn, D.C.; Chatterley, T.; Dorgan, M.; Lok, C.E.; Fitchett, D.; Tsuyuki, R.T. Effect of calcium-based versus non-calcium-based phosphate binders on mortality in patients with chronic kidney disease: An updated systematic review and meta-analysis. Lancet 2013, 382, 1268–1277. [Google Scholar] [CrossRef]
- Ruospo, M.; Palmer, S.C.; Natale, P.; Craig, J.C.; Vecchio, M.; Elder, G.J.; Strippoli, G.F. Phosphate binders for preventing and treating chronic kidney disease-mineral and bone disorder (CKD-MBD). Cochrane Database Syst. Rev. 2018, 8, CD006023. [Google Scholar] [CrossRef]
- Block, G.A.; Bushinsky, D.A.; Cheng, S.; Cunningham, J.; Dehmel, B.; Drueke, T.B.; Ketteler, M.; Kewalramani, R.; Martin, K.J.; Moe, S.M.; et al. Effect of Etelcalcetide vs Cinacalcet on Serum Parathyroid Hormone in Patients Receiving Hemodialysis With Secondary Hyperparathyroidism: A Randomized Clinical Trial. JAMA 2017, 317, 156–164. [Google Scholar] [CrossRef]
- Wu, M.; Tang, R.N.; Liu, H.; Pan, M.M.; Liu, B.C. Cinacalcet ameliorates aortic calcification in uremic rats via suppression of endothelial-to-mesenchymal transition. Acta Pharmacol. Sin. 2016, 37, 1423–1431. [Google Scholar] [CrossRef] [Green Version]
- Friedl, C.; Zitt, E. Role of etelcalcetide in the management of secondary hyperparathyroidism in hemodialysis patients: A review on current data and place in therapy. Drug Des. Dev. Ther. 2018, 12, 1589–1598. [Google Scholar] [CrossRef] [Green Version]
- Investigators, E.T.; Chertow, G.M.; Block, G.A.; Correa-Rotter, R.; Drueke, T.B.; Floege, J.; Goodman, W.G.; Herzog, C.A.; Kubo, Y.; London, G.M.; et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N. Engl. J. Med. 2012, 367, 2482–2494. [Google Scholar] [CrossRef] [Green Version]
- Raggi, P.; Chertow, G.M.; Torres, P.U.; Csiky, B.; Naso, A.; Nossuli, K.; Moustafa, M.; Goodman, W.G.; Lopez, N.; Downey, G.; et al. The ADVANCE study: A randomized study to evaluate the effects of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol. Dial. Transplant. 2011, 26, 1327–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, S.C.; Nistor, I.; Craig, J.C.; Pellegrini, F.; Messa, P.; Tonelli, M.; Covic, A.; Strippoli, G.F. Cinacalcet in patients with chronic kidney disease: A cumulative meta-analysis of randomized controlled trials. PLoS Med. 2013, 10, e1001436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, M.; Methven, S.; Gasparini, A.; Barany, P.; Birnie, K.; MacNeill, S.; May, M.T.; Caskey, F.J.; Carrero, J.J. Cinacalcet use and the risk of cardiovascular events, fractures and mortality in chronic kidney disease patients with secondary hyperparathyroidism. Sci. Rep. 2018, 8, 2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Li, M.; You, L.; Li, H.; Ni, L.; Gu, Y.; Hao, C.; Chen, J. Effects and safety of calcimimetics in end stage renal disease patients with secondary hyperparathyroidism: A meta-analysis. PLoS ONE 2012, 7, e48070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toussaint, N.D.; Elder, G.J.; Kerr, P.G. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin. J. Am. Soc. Nephrol. 2009, 4, 221–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perazella, M.A.; Markowitz, G.S. Bisphosphonate nephrotoxicity. Kidney Int. 2008, 74, 1385–1393. [Google Scholar] [CrossRef] [Green Version]
- Bergner, R.; Diel, I.J.; Henrich, D.; Hoffmann, M.; Uppenkamp, M. Differences in nephrotoxicity of intravenous bisphosphonates for the treatment of malignancy-related bone disease. Onkologie 2006, 29, 534–540. [Google Scholar] [CrossRef]
- Markowitz, G.S.; Appel, G.B.; Fine, P.L.; Fenves, A.Z.; Loon, N.R.; Jagannath, S.; Kuhn, J.A.; Dratch, A.D.; D’Agati, V.D. Collapsing focal segmental glomerulosclerosis following treatment with high-dose pamidronate. J. Am. Soc. Nephrol. 2001, 12, 1164–1172. [Google Scholar]
- Verhulst, A.; Sun, S.; McKenna, C.E.; D’Haese, P.C. Endocytotic uptake of zoledronic acid by tubular cells may explain its renal effects in cancer patients receiving high doses of the compound. PLoS ONE 2015, 10, e0121861. [Google Scholar] [CrossRef] [Green Version]
- Otto, S.; Pautke, C.; Hafner, S.; Hesse, R.; Reichardt, L.F.; Mast, G.; Ehrenfeld, M.; Cornelius, C.P. Pathologic fractures in bisphosphonate-related osteonecrosis of the jaw-review of the literature and review of our own cases. Craniomaxillofac. Trauma Reconstr. 2013, 6, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Miller, P.D.; Roux, C.; Boonen, S.; Barton, I.P.; Dunlap, L.E.; Burgio, D.E. Safety and efficacy of risedronate in patients with age-related reduced renal function as estimated by the Cockcroft and Gault method: A pooled analysis of nine clinical trials. J. Bone Miner. Res. 2005, 20, 2105–2115. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J. Bisphosphonates in the renal patient. Nephrol. Dial. Transplant. 2007, 22, 1505–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bostrom, K.I.; Rajamannan, N.M.; Towler, D.A. The regulation of valvular and vascular sclerosis by osteogenic morphogens. Circ. Res. 2011, 109, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Suzuki, Y.; Matsushita, M.; Fujii, H.; Miyaura, C.; Aizawa, S.; Kogo, H. Prevention of aortic calcification by etidronate in the renal failure rat model. Eur. J. Pharmacol. 2007, 558, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Caffarelli, C.; Montagnani, A.; Nuti, R.; Gonnelli, S. Bisphosphonates, atherosclerosis and vascular calcification: Update and systematic review of clinical studies. Clin. Interv. Aging 2017, 12, 1819–1828. [Google Scholar] [CrossRef] [Green Version]
- Toussaint, N.D.; Lau, K.K.; Strauss, B.J.; Polkinghorne, K.R.; Kerr, P.G. Effect of alendronate on vascular calcification in CKD stages 3 and 4: A pilot randomized controlled trial. Am. J. Kidney Dis. 2010, 56, 57–68. [Google Scholar] [CrossRef]
- Hartle, J.E.; Tang, X.; Kirchner, H.L.; Bucaloiu, I.D.; Sartorius, J.A.; Pogrebnaya, Z.V.; Akers, G.A.; Carnero, G.E.; Perkins, R.M. Bisphosphonate therapy, death, and cardiovascular events among female patients with CKD: A retrospective cohort study. Am. J. Kidney Dis. 2012, 59, 636–644. [Google Scholar] [CrossRef]
- Kranenburg, G.; Bartstra, J.W.; Weijmans, M.; de Jong, P.A.; Mali, W.P.; Verhaar, H.J.; Visseren, F.L.J.; Spiering, W. Bisphosphonates for cardiovascular risk reduction: A systematic review and meta-analysis. Atherosclerosis 2016, 252, 106–115. [Google Scholar] [CrossRef]
- McCloskey, E.V.; Johansson, H.; Oden, A.; Austin, M.; Siris, E.; Wang, A.; Lewiecki, E.M.; Lorenc, R.; Libanati, C.; Kanis, J.A. Denosumab reduces the risk of osteoporotic fractures in postmenopausal women, particularly in those with moderate to high fracture risk as assessed with FRAX. J. Bone Miner. Res. 2012, 27, 1480–1486. [Google Scholar] [CrossRef]
- Feng, X.; Teitelbaum, S.L. Osteoclasts: New Insights. Bone Res. 2013, 1, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Panizo, S.; Cardus, A.; Encinas, M.; Parisi, E.; Valcheva, P.; Lopez-Ongil, S.; Coll, B.; Fernandez, E.; Valdivielso, J.M. RANKL increases vascular smooth muscle cell calcification through a RANK-BMP4-dependent pathway. Circ. Res. 2009, 104, 1041–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helas, S.; Goettsch, C.; Schoppet, M.; Zeitz, U.; Hempel, U.; Morawietz, H.; Kostenuik, P.J.; Erben, R.G.; Hofbauer, L.C. Inhibition of receptor activator of NF-kappaB ligand by denosumab attenuates vascular calcium deposition in mice. Am. J. Pathol. 2009, 175, 473–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iseri, K.; Watanabe, M.; Yoshikawa, H.; Mitsui, H.; Endo, T.; Yamamoto, Y.; Iyoda, M.; Ryu, K.; Inaba, T.; Shibata, T.; et al. Effects of Denosumab and Alendronate on Bone Health and Vascular Function in Hemodialysis Patients: A Randomized, Controlled Trial. J. Bone Miner. Res. 2019, 34, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Hjortnaes, J.; Bouten, C.V.; Van Herwerden, L.A.; Grundeman, P.F.; Kluin, J. Translating autologous heart valve tissue engineering from bench to bed. Tissue Eng. Part B Rev. 2009, 15, 307–317. [Google Scholar] [CrossRef]
- Rajamannan, N.M.; Evans, F.J.; Aikawa, E.; Grande-Allen, K.J.; Demer, L.L.; Heistad, D.D.; Simmons, C.A.; Masters, K.S.; Mathieu, P.; O’Brien, K.D.; et al. Calcific aortic valve disease: Not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation 2011, 124, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- An, W.S.; Lee, S.M.; Son, Y.K.; Kim, S.E. Combination of omega-3 fatty acid and menaquinone-7 prevents progression of aortic calcification in adenine and low protein diet induced rat model. Nephrol. Dial. Transplant. 2017, 32, iii253–iii254. [Google Scholar] [CrossRef]
- Diaz-Tocados, J.M.; Peralta-Ramirez, A.; Rodriguez-Ortiz, M.E.; Raya, A.I.; Lopez, I.; Pineda, C.; Herencia, C.; Montes de Oca, A.; Vergara, N.; Steppan, S.; et al. Dietary magnesium supplementation prevents and reverses vascular and soft tissue calcifications in uremic rats. Kidney Int. 2017, 92, 1084–1099. [Google Scholar] [CrossRef]
- Kaesler, N.; Goettsch, C.; Weis, D.; Schurgers, L.; Hellmann, B.; Floege, J.; Kramann, R. Magnesium but not nicotinamide prevents vascular calcification in experimental uraemia. Nephrol. Dial. Transplant. 2019. [Google Scholar] [CrossRef]
- O’Young, J.; Liao, Y.; Xiao, Y.; Jalkanen, J.; Lajoie, G.; Karttunen, M.; Goldberg, H.A.; Hunter, G.K. Matrix Gla protein inhibits ectopic calcification by a direct interaction with hydroxyapatite crystals. J. Am. Chem. Soc. 2011, 133, 18406–18412. [Google Scholar] [CrossRef]
- Tesfamariam, B. Involvement of Vitamin K-Dependent Proteins in Vascular Calcification. J. Cardiovasc. Pharmacol. Ther. 2019, 24, 323–333. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Teunissen, K.J.; Knapen, M.H.; Kwaijtaal, M.; van Diest, R.; Appels, A.; Reutelingsperger, C.P.; Cleutjens, J.P.; Vermeer, C. Novel conformation-specific antibodies against matrix gamma-carboxyglutamic acid (Gla) protein: Undercarboxylated matrix Gla protein as marker for vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1629–1633. [Google Scholar] [CrossRef] [Green Version]
- Halder, M.; Petsophonsakul, P.; Akbulut, A.C.; Pavlic, A.; Bohan, F.; Anderson, E.; Maresz, K.; Kramann, R.; Schurgers, L. Vitamin K: Double Bonds beyond Coagulation Insights into Differences between Vitamin K1 and K2 in Health and Disease. Int. J. Mol. Sci. 2019, 20, 896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cranenburg, E.C.; Schurgers, L.J.; Uiterwijk, H.H.; Beulens, J.W.; Dalmeijer, G.W.; Westerhuis, R.; Magdeleyns, E.J.; Herfs, M.; Vermeer, C.; Laverman, G.D.; et al. Vitamin K intake and status are low in hemodialysis patients. Kidney Int. 2012, 82, 605–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.M.; An, W.S. Supplementary nutrients for prevention of vascular calcification in patients with chronic kidney disease. Korean J. Intern. Med. 2019, 34, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Schlieper, G.; Westenfeld, R.; Kruger, T.; Cranenburg, E.C.; Magdeleyns, E.J.; Brandenburg, V.M.; Djuric, Z.; Damjanovic, T.; Ketteler, M.; Vermeer, C.; et al. Circulating nonphosphorylated carboxylated matrix gla protein predicts survival in ESRD. J. Am. Soc. Nephrol. 2011, 22, 387–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyzer, C.A.; de Borst, M.H.; van den Berg, E.; Jahnen-Dechent, W.; Arampatzis, S.; Farese, S.; Bergmann, I.P.; Floege, J.; Navis, G.; Bakker, S.J.; et al. Calcification Propensity and Survival among Renal Transplant Recipients. J. Am. Soc. Nephrol. 2016, 27, 239–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, K.; Masuda, K.; Yamazaki, M.; Kiyohara, C.; Itoh, S.; Wasaki, M.; Inoue, H. Metal ion and vitamin adsorption profiles of phosphate binder ion-exchange resins. Clin. Nephrol. 2010, 73, 30–35. [Google Scholar] [CrossRef]
- Neradova, A.; Schumacher, S.P.; Hubeek, I.; Lux, P.; Schurgers, L.J.; Vervloet, M.G. Phosphate binders affect vitamin K concentration by undesired binding, an in vitro study. BMC Nephrol. 2017, 18, 149. [Google Scholar] [CrossRef]
- Jansz, T.T.; Neradova, A.; van Ballegooijen, A.J.; Verhaar, M.C.; Vervloet, M.G.; Schurgers, L.J.; van Jaarsveld, B.C. The role of kidney transplantation and phosphate binder use in vitamin K status. PLoS ONE 2018, 13, e0203157. [Google Scholar] [CrossRef]
- Aoun, M.; Makki, M.; Azar, H.; Matta, H.; Chelala, D.N. High Dephosphorylated-Uncarboxylated MGP in Hemodialysis patients: Risk factors and response to vitamin K2, A pre-post intervention clinical trial. BMC Nephrol. 2017, 18, 191. [Google Scholar] [CrossRef] [PubMed]
- Westenfeld, R.; Krueger, T.; Schlieper, G.; Cranenburg, E.C.; Magdeleyns, E.J.; Heidenreich, S.; Holzmann, S.; Vermeer, C.; Jahnen-Dechent, W.; Ketteler, M.; et al. Effect of vitamin K2 supplementation on functional vitamin K deficiency in hemodialysis patients: A randomized trial. Am. J. Kidney Dis. 2012, 59, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Caluwe, R.; Pyfferoen, L.; De Boeck, K.; De Vriese, A.S. The effects of vitamin K supplementation and vitamin K antagonists on progression of vascular calcification: Ongoing randomized controlled trials. Clin. Kidney J. 2016, 9, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Kurnatowska, I.; Grzelak, P.; Masajtis-Zagajewska, A.; Kaczmarska, M.; Stefanczyk, L.; Vermeer, C.; Maresz, K.; Nowicki, M. Effect of vitamin K2 on progression of atherosclerosis and vascular calcification in nondialyzed patients with chronic kidney disease stages 3–5. Pol. Arch. Med. Wewn. 2015, 125, 631–640. [Google Scholar] [CrossRef]
- Zheng, Z.; Shi, H.; Jia, J.; Li, D.; Lin, S. Vitamin D supplementation and mortality risk in chronic kidney disease: A meta-analysis of 20 observational studies. BMC Nephrol. 2013, 14, 199. [Google Scholar] [CrossRef] [Green Version]
- Chitalia, N.; Recio-Mayoral, A.; Kaski, J.C.; Banerjee, D. Vitamin D deficiency and endothelial dysfunction in non-dialysis chronic kidney disease patients. Atherosclerosis 2012, 220, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.; Tang, M.; Perry, T.; Zalunardo, N.; Beaulieu, M.; Dubland, J.A.; Zerr, K.; Djurdjev, O. Randomized Controlled Trial for the Effect of Vitamin D Supplementation on Vascular Stiffness in CKD. Clin. J. Am. Soc. Nephrol. 2017, 12, 1447–1460. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Yadav, A.K.; Lal, A.; Kumar, V.; Singhal, M.; Billot, L.; Gupta, K.L.; Banerjee, D.; Jha, V. A Randomized Trial of Vitamin D Supplementation on Vascular Function in CKD. J. Am. Soc. Nephrol. 2017, 28, 3100–3108. [Google Scholar] [CrossRef] [Green Version]
- Samaan, F.; Carvalho, A.B.; Pillar, R.; Rocha, L.A.; Cassiolato, J.L.; Cuppari, L.; Canziani, M.E.F. The Effect of Long-Term Cholecalciferol Supplementation on Vascular Calcification in Chronic Kidney Disease Patients With Hypovitaminosis D. J. Ren. Nutr. 2019, 29, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Levin, A.; Le Barbier, M.; Er, L.; Andress, D.; Sigrist, M.K.; Djurdjev, O. Incident isolated 1,25(OH)(2)D(3) deficiency is more common than 25(OH)D deficiency in CKD. J. Nephrol. 2012, 25, 204–210. [Google Scholar] [CrossRef]
- Louvet, L.; Buchel, J.; Steppan, S.; Passlick-Deetjen, J.; Massy, Z.A. Magnesium prevents phosphate-induced calcification in human aortic vascular smooth muscle cells. Nephrol. Dial. Transplant. 2013, 28, 869–878. [Google Scholar] [CrossRef] [Green Version]
- Massy, Z.A.; Drueke, T.B. Magnesium and outcomes in patients with chronic kidney disease: Focus on vascular calcification, atherosclerosis and survival. Clin. Kidney J. 2012, 5, i52–i61. [Google Scholar] [CrossRef] [Green Version]
- Briet, M.; Schiffrin, E.L. Vascular actions of aldosterone. J. Vasc. Res. 2013, 50, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Mizuiri, S.; Nishizawa, Y.; Yamashita, K.; Naito, T.; Ono, K.; Tanji, C.; Usui, K.; Doi, S.; Masaki, T.; Shigemoto, K. Hypomagnesemia is not an independent risk factor for mortality in Japanese maintenance hemodialysis patients. Int. Urol. Nephrol. 2019, 51, 1043–1052. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Cai, K.; Luo, Q.; Wang, L.; Hong, Y. Baseline Serum Magnesium Level and Its Variability in Maintenance Hemodialysis Patients: Associations with Mortality. Kidney Blood Press. Res. 2019, 44, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, Y.; Fujii, N.; Shoji, T.; Hayashi, T.; Rakugi, H.; Iseki, K.; Tsubakihara, Y.; Isaka, Y.; Committee of Renal Data Registry of the Japanese Society for Dialysis Therapy. Magnesium modifies the cardiovascular mortality risk associated with hyperphosphatemia in patients undergoing hemodialysis: A cohort study. PLoS ONE 2014, 9, e116273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, K.; Luo, Q.; Dai, Z.; Zhu, B.; Fei, J.; Xue, C.; Wu, D. Hypomagnesemia Is Associated with Increased Mortality among Peritoneal Dialysis Patients. PLoS ONE 2016, 11, e0152488. [Google Scholar] [CrossRef] [Green Version]
- Bundy, J.D.; Cai, X.; Scialla, J.J.; Dobre, M.A.; Chen, J.; Hsu, C.Y.; Leonard, M.B.; Go, A.S.; Rao, P.S.; Lash, J.P.; et al. Serum Calcification Propensity and Coronary Artery Calcification Among Patients With CKD: The CRIC (Chronic Renal Insufficiency Cohort) Study. Am. J. Kidney Dis. 2019, 73, 806–814. [Google Scholar] [CrossRef]
- Mortazavi, M.; Moeinzadeh, F.; Saadatnia, M.; Shahidi, S.; McGee, J.C.; Minagar, A. Effect of magnesium supplementation on carotid intima-media thickness and flow-mediated dilatation among hemodialysis patients: A double-blind, randomized, placebo-controlled trial. Eur. Neurol. 2013, 69, 309–316. [Google Scholar] [CrossRef]
- Turgut, F.; Kanbay, M.; Metin, M.R.; Uz, E.; Akcay, A.; Covic, A. Magnesium supplementation helps to improve carotid intima media thickness in patients on hemodialysis. Int. Urol. Nephrol. 2008, 40, 1075–1082. [Google Scholar] [CrossRef]
- Bressendorff, I.; Hansen, D.; Schou, M.; Kragelund, C.; Brandi, L. The effect of magnesium supplementation on vascular calcification in chronic kidney disease-a randomised clinical trial (MAGiCAL-CKD): Essential study design and rationale. BMJ Open 2017, 7, e016795. [Google Scholar] [CrossRef]
- Salcedo, C.; Joubert, P.H.; Ferrer, M.D.; Canals, A.Z.; Maduell, F.; Torregrosa, V.; Campistol, J.M.; Ojeda, R.; Perello, J. A phase 1b randomized, placebo-controlled clinical trial with SNF472 in haemodialysis patients. Br. J. Clin. Pharmacol. 2019, 85, 796–806. [Google Scholar] [CrossRef] [Green Version]
- Perello, J.; Joubert, P.H.; Ferrer, M.D.; Canals, A.Z.; Sinha, S.; Salcedo, C. First-time-in-human randomized clinical trial in healthy volunteers and haemodialysis patients with SNF472, a novel inhibitor of vascular calcification. Br. J. Clin. Pharmacol. 2018, 84, 2867–2876. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.X.; Zhang, H.; Peng, C. Puerarin: A review of pharmacological effects. Phytother. Res. 2014, 28, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, X.; Zhong, X.; Li, Z.; Cai, S.; Yang, P.; Ou, C.; Chen, M. Puerarin inhibits vascular calcification of uremic rats. Eur. J. Pharmacol. 2019, 855, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Millar, J.S. Novel benefits of peroxisome proliferator-activated receptors on cardiovascular risk. Curr. Opin. Lipidol. 2013, 24, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Ikejima, H.; Imanishi, T.; Tsujioka, H.; Kuroi, A.; Kobayashi, K.; Shiomi, M.; Muragaki, Y.; Mochizuki, S.; Goto, M.; Yoshida, K.; et al. Effects of telmisartan, a unique angiotensin receptor blocker with selective peroxisome proliferator-activated receptor-gamma-modulating activity, on nitric oxide bioavailability and atherosclerotic change. J. Hypertens. 2008, 26, 964–972. [Google Scholar] [CrossRef]
- Bostom, A.; Pasch, A.; Madsen, T.; Roberts, M.B.; Franceschini, N.; Steubl, D.; Garimella, P.S.; Ix, J.H.; Tuttle, K.R.; Ivanova, A.; et al. Serum Calcification Propensity and Fetuin-A: Biomarkers of Cardiovascular Disease in Kidney Transplant Recipients. Am. J. Nephrol. 2018, 48, 21–31. [Google Scholar] [CrossRef]
- Zhao, G.; Xu, M.J.; Zhao, M.M.; Dai, X.Y.; Kong, W.; Wilson, G.M.; Guan, Y.; Wang, C.Y.; Wang, X. Activation of nuclear factor-kappa B accelerates vascular calcification by inhibiting ankylosis protein homolog expression. Kidney Int. 2012, 82, 34–44. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Yamashita, M.; Horimai, C.; Hayashi, M. Smooth Muscle-Selective Nuclear Factor-kappaB Inhibition Reduces Phosphate-Induced Arterial Medial Calcification in Mice With Chronic Kidney Disease. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Taniguchi, M.; Tokumoto, M.; Toyonaga, J.; Fujisaki, K.; Suehiro, T.; Noguchi, H.; Iida, M.; Tsuruya, K.; Kitazono, T. The antioxidant tempol ameliorates arterial medial calcification in uremic rats: Important role of oxidative stress in the pathogenesis of vascular calcification in chronic kidney disease. J. Bone Miner. Res. 2012, 27, 474–485. [Google Scholar] [CrossRef]
- Tatsumoto, N.; Yamada, S.; Tokumoto, M.; Eriguchi, M.; Noguchi, H.; Torisu, K.; Tsuruya, K.; Kitazono, T. Spironolactone ameliorates arterial medial calcification in uremic rats: The role of mineralocorticoid receptor signaling in vascular calcification. Am. J. Physiol. Ren. Physiol. 2015, 309, F967–F979. [Google Scholar] [CrossRef] [Green Version]
- Scialla, J.J.; Kao, W.H.; Crainiceanu, C.; Sozio, S.M.; Oberai, P.C.; Shafi, T.; Coresh, J.; Powe, N.R.; Plantinga, L.C.; Jaar, B.G.; et al. Biomarkers of vascular calcification and mortality in patients with ESRD. Clin. J. Am. Soc. Nephrol. 2014, 9, 745–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Lu, R.; Zhang, M.; Pang, H.; Zhu, M.; Zhang, W.; Ni, Z.; Qian, J.; Yan, Y. Serum Soluble Klotho Level Is Associated with Abdominal Aortic Calcification in Patients on Maintenance Hemodialysis. Blood Purif. 2015, 40, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Pasch, A.; Farese, S.; Graber, S.; Wald, J.; Richtering, W.; Floege, J.; Jahnen-Dechent, W. Nanoparticle-based test measures overall propensity for calcification in serum. J. Am. Soc. Nephrol. 2012, 23, 1744–1752. [Google Scholar] [CrossRef] [PubMed]
- Heiss, A.; DuChesne, A.; Denecke, B.; Grotzinger, J.; Yamamoto, K.; Renne, T.; Jahnen-Dechent, W. Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J. Biol. Chem. 2003, 278, 13333–13341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.R.; Ford, M.L.; Tomlinson, L.A.; Bodenham, E.; McMahon, L.P.; Farese, S.; Rajkumar, C.; Holt, S.G.; Pasch, A. Serum calcification propensity predicts all-cause mortality in predialysis CKD. J. Am. Soc. Nephrol. 2014, 25, 339–348. [Google Scholar] [CrossRef]
- Lorenz, G.; Steubl, D.; Kemmner, S.; Pasch, A.; Koch-Sembdner, W.; Pham, D.; Haller, B.; Bachmann, Q.; Mayer, C.C.; Wassertheurer, S.; et al. Worsening calcification propensity precedes all-cause and cardiovascular mortality in haemodialyzed patients. Sci. Rep. 2017, 7, 13368. [Google Scholar] [CrossRef] [Green Version]
- Dahle, D.O.; Asberg, A.; Hartmann, A.; Holdaas, H.; Bachtler, M.; Jenssen, T.G.; Dionisi, M.; Pasch, A. Serum Calcification Propensity Is a Strong and Independent Determinant of Cardiac and All-Cause Mortality in Kidney Transplant Recipients. Am. J. Transplant. 2016, 16, 204–212. [Google Scholar] [CrossRef] [Green Version]
- Brandenburg, V.M.; Reinartz, S.; Kaesler, N.; Kruger, T.; Dirrichs, T.; Kramann, R.; Peeters, F.; Floege, J.; Keszei, A.; Marx, N.; et al. Slower Progress of Aortic Valve Calcification With Vitamin K Supplementation: Results From a Prospective Interventional Proof-of-Concept Study. Circulation 2017, 135, 2081–2083. [Google Scholar] [CrossRef]



| Treatment | Substance | Dosis | Medication | Experimental Model | Species, Strain | Ref. |
|---|---|---|---|---|---|---|
| Phosphate binder | Sevelamer | 750 mg/kg | Daily oral gavage, 4 weeks | Adenine diet | Wistar rat | [32] |
| Phosphate binder | Sevelamer | 3% | Diet, 6 months | 5/6 nephrectomy | Sprague-Dawley rat | [31] |
| Phosphate binder | CaMg | 185 mg/kg | Daily oral gavage, 6 weeks | Adenine diet | Wistar rat | [33] |
| Calcimimetic | Cinacalcet | 10 mg/kg | Daily oral gavage, 12 weeks | Adenine diet | Wistar rat | [38] |
| Patients | Follow-up | Main Results | Ref. |
|---|---|---|---|
| CKD stages 4 to 5D (n = 107) | 2.2 years | dp-ucMGP: positive association with progressive CKD stages and increased all-cause mortality | [75] |
| HD patients (n = 188) | 3 years | - 6.5-fold elevated dp-ucMGP - dp-cMGP associated with increased all-cause and CV mortality | [75] |
| KTR (n = 518) | 9.8 years | dp-ucMGP: association with increased all-cause mortality | [76] |
| Patients | Treatment | Study Design | Main Results | Ref. |
|---|---|---|---|---|
| CKD stage 3–5 (n = 42) | 90 μg/d MK-7 + 10 μg/d cholecalciferol, or 10 μg/d cholecalciferol (control), 38.5 weeks | Prospective, randomized, double-blind | Decrease of dp-ucMGP, smaller increase of CAC and CCA-IMT compared to control | [83] |
| HD patients (n = 50) | 360 μg/d MK-7, 4 weeks | Prospective, pre-post intervention clinical trial | 86% decrease of dp-ucMGP | [80] |
| HD patients (n = 17) | 135 μg/d MK-7, 6 weeks | Interventional pilot study | Decrease of dp-ucMGP but not dp-cMGP | [75] |
| HD patients (n = 53), Healthy controls (n = 50) | 45, 135, 360 μg/d MK-7, 6 weeks | Interventional, randomized, non-placebo-controlled trial | Dose-dependent decrease of dp-ucMGP | [81] |
| Patients | Mean/Median T50 (Baseline) | Follow up, Years | Findings | Ref. |
|---|---|---|---|---|
| CKD stages 2 to 4 (n = 1274), In follow up n = 780 | Median: 321 min | 3.2 | Association of low T50 with increased CAC prevalence and progression | [97] |
| CKD stages 3 and 4 (n = 184) | Mean: 329 ± 95 min | 5.3 | Association of low T50 with increased all-cause mortality and APWV | [116] |
| HD patients (n = 2785), control group (n = 1366) | Mean: 212 min (10th–90th percentile: 109–328 min) | 1.7 | Association of low T50 with increased all-cause mortality and CVD | [117] |
| HD patients (n = 188) | Mean: 246 ± 64 min | 3.7 | Association of low T50 and T50 decline with all-cause and CV mortality | [117] |
| KTR (n = 699) | Mean: 286 ± 62 min | 3.1 | Association of low T50 with increased all-cause and CV mortality and graft failure | [76] |
| KTR (n = 433) | Mean: 340 ± 70 min | 3.7 | Association of low T50 with increased CVD event risk | [107] |
| KTR during 10 weeks after transplantation (n = 1435), Follow-up: APWV after 1 year (n = 589) | Median: 188 min (25th–75th percentile: 139–248 min) | 5.1 | Association of low T50 with increased all-cause and CV mortality APWV not associated with T50 baseline | [118] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Himmelsbach, A.; Ciliox, C.; Goettsch, C. Cardiovascular Calcification in Chronic Kidney Disease—Therapeutic Opportunities. Toxins 2020, 12, 181. https://doi.org/10.3390/toxins12030181
Himmelsbach A, Ciliox C, Goettsch C. Cardiovascular Calcification in Chronic Kidney Disease—Therapeutic Opportunities. Toxins. 2020; 12(3):181. https://doi.org/10.3390/toxins12030181
Chicago/Turabian StyleHimmelsbach, Anika, Carina Ciliox, and Claudia Goettsch. 2020. "Cardiovascular Calcification in Chronic Kidney Disease—Therapeutic Opportunities" Toxins 12, no. 3: 181. https://doi.org/10.3390/toxins12030181
APA StyleHimmelsbach, A., Ciliox, C., & Goettsch, C. (2020). Cardiovascular Calcification in Chronic Kidney Disease—Therapeutic Opportunities. Toxins, 12(3), 181. https://doi.org/10.3390/toxins12030181