Potential Roles and Functions of Listerial Virulence Factors during Brain Entry


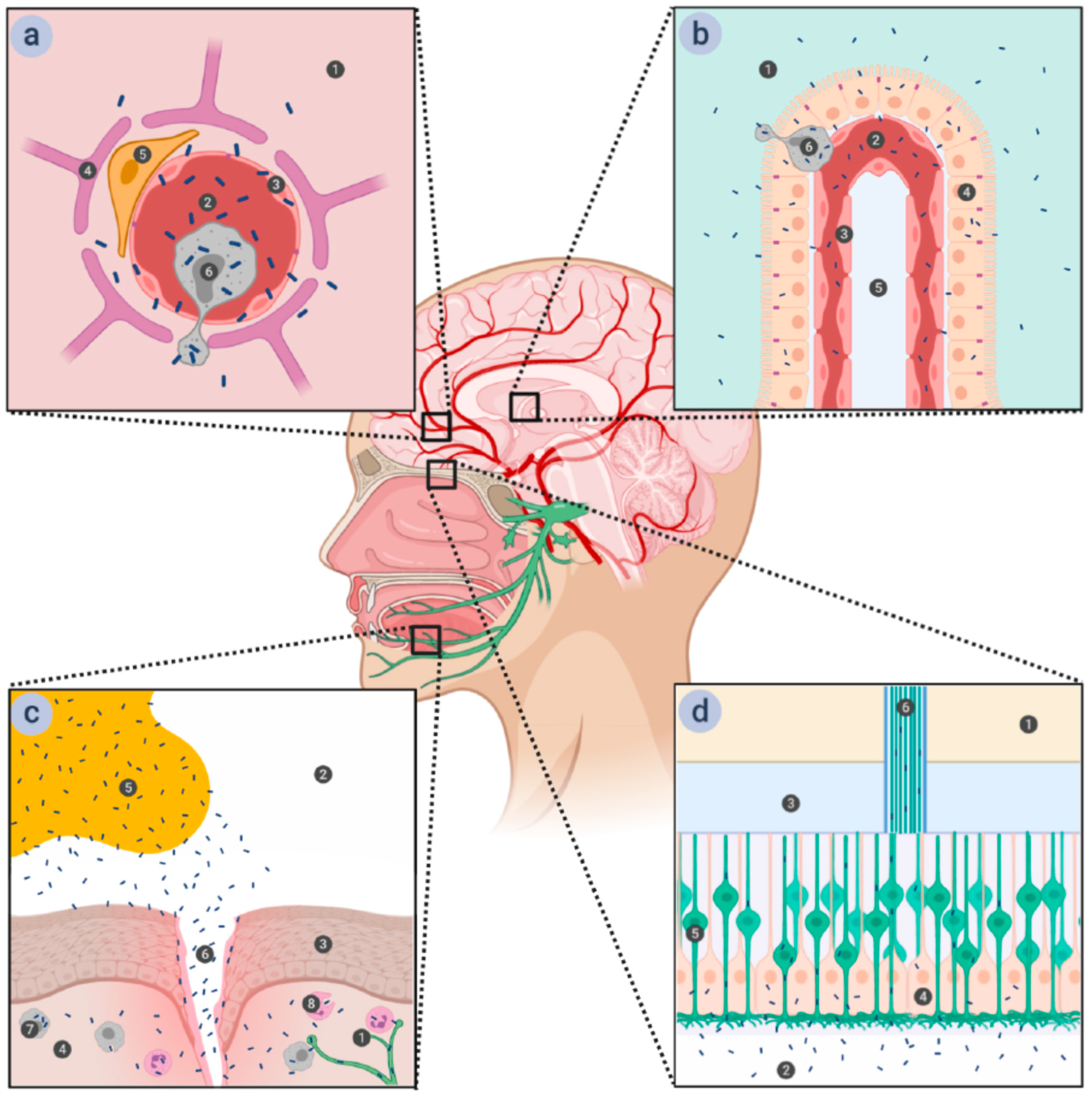{kind=link}
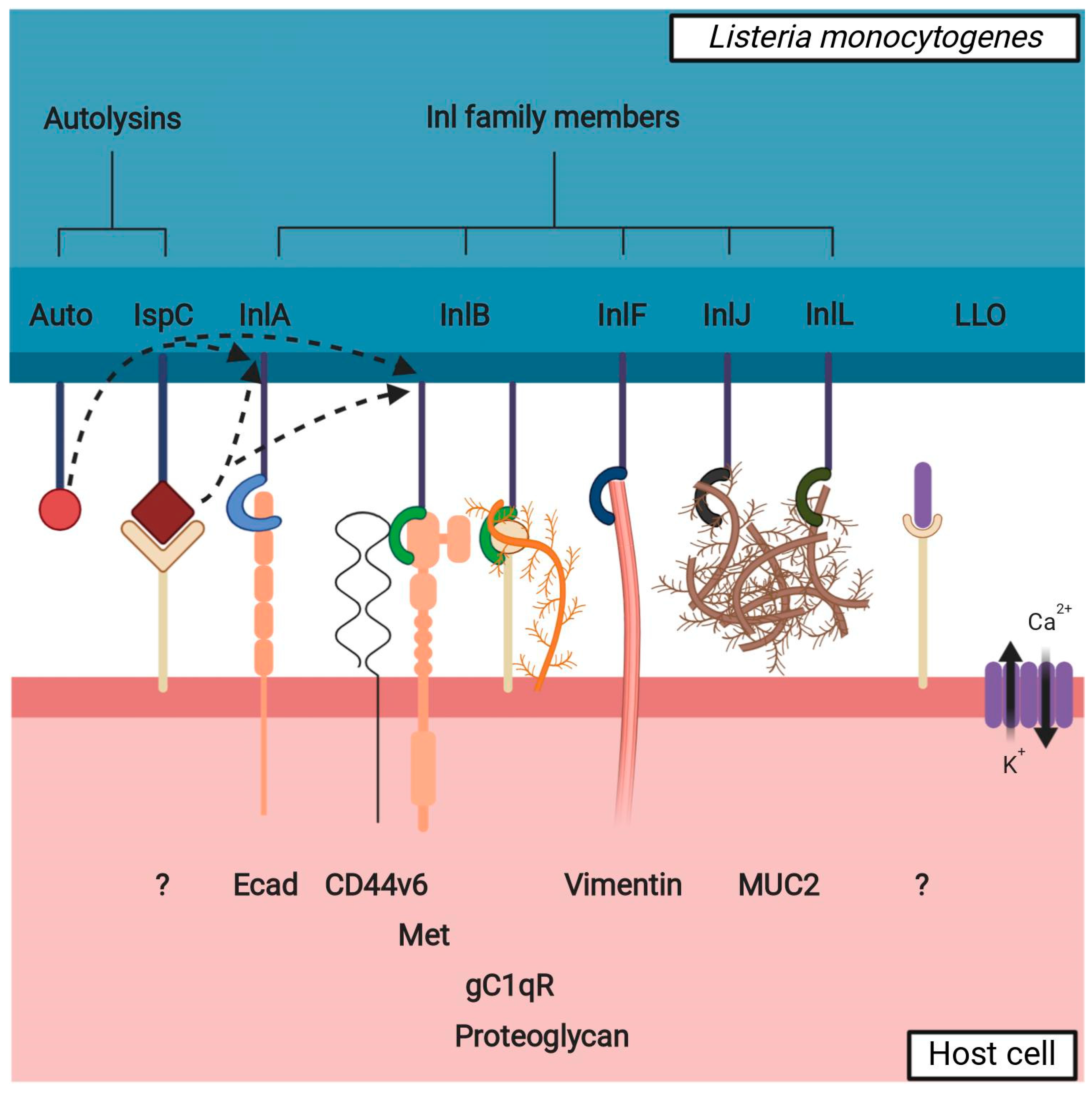{kind=link}
Abstract
:1. Introduction
2. Virulence Factors
2.1. Internalins
2.1.1. Internalin (InlA)
2.1.2. Internalin B (InlB)
2.1.3. Internalin F (InlF)
2.1.4. Internalin J (InlJ)
2.1.5. Internalin L (InlL)
2.2. Other Virulence Factors
2.2.1. Auto
2.2.2. IspC
2.2.3. Listeriolysin O (LLO)
2.2.4. Proteins Encoded by Listerial Pathogenicity Island 4 (LIPI-4)
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Radoshevich, L.; Cossart, P. Listeria monocytogenes: Towards a complete picture of its physiology and pathogenesis. Nat. Rev. Microbiol. 2018, 16, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Maury, M.M.; Tsai, Y.H.; Charlier, C.; Touchon, M.; Chenal-Francisque, V.; Leclercq, A.; Criscuolo, A.; Gautier, C.; Roussel, S.; Brisabois, A.; et al. Uncovering Listeria monocytogenes hypervirulence by harnessing its biodiversity. Nat. Genet. 2016, 48, 308–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiden, M.C.; Bygraves, J.A.; Feil, E.; Morelli, G.; Russell, J.E.; Urwin, R.; Zhang, Q.; Zhou, J.; Zurth, K.; Caugant, D.A.; et al. Multilocus sequence typing: A portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad Sci. USA 1998, 95, 3140–3145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragon, M.; Wirth, T.; Hollandt, F.; Lavenir, R.; Lecuit, M.; Le Monnier, A.; Brisse, S. A new perspective on Listeria monocytogenes evolution. PLoS Pathog. 2008, 4, e1000146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierne, H.; Milohanic, E.; Kortebi, M. To be cytosolic or vacuolar: The double life of listeria monocytogenes. Front. Cell Infect. Microbiol. 2018, 8, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlech, W.F., 3rd; Lavigne, P.M.; Bortolussi, R.A.; Allen, A.C.; Haldane, E.V.; Wort, A.J.; Hightower, A.W.; Johnson, S.E.; King, S.H.; Nicholls, E.S.; et al. Epidemic listeriosis—Evidence for transmission by food. New Engl. J. Med. 1983, 308, 203–206. [Google Scholar] [CrossRef]
- Drevets, D.A.; Bronze, M.S. Listeria monocytogenes: Epidemiology, human disease, and mechanisms of brain invasion. FEMS Immunol. Med. Microbiol. 2008, 53, 151–165. [Google Scholar] [CrossRef] [Green Version]
- Drevets, D.A. Dissemination of Listeria monocytogenes by infected phagocytes. Infect. Immun. 1999, 67, 3512–3517. [Google Scholar] [CrossRef] [Green Version]
- Dando, S.J.; Mackay-Sim, A.; Norton, R.; Currie, B.J.; St. John, J.A.; Ekberg, J.A.K.; Batzloff, M.; Ulett, G.C.; Beacham, I.R. Pathogens penetrating the central nervous system: Infection pathways and the cellular and molecular mechanisms of invasion. Clin. Microbiol. Rev. 2014, 27, 691–726. [Google Scholar] [CrossRef] [Green Version]
- Berche, P. Bacteremia is required for invasion of the murine central nervous system by Listeria monocytogenes. Microb. Pathog. 1995, 18, 323–336. [Google Scholar] [CrossRef]
- Greiffenberg, L.; Goebel, W.; Kim, K.S.; Weiglein, I.; Bubert, A.; Engelbrecht, F.; Stins, M.; Kuhn, M. Interaction of Listeria monocytogenes with human brain microvascular endothelial cells: InlB-dependent invasion, long-term intracellular growth, and spread from macrophages to endothelial cells. Infect. Immun. 1998, 66, 5260–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drevets, D.A.; Jelinek, T.A.; Freitag, N.E. Listeria monocytogenes-infected phagocytes can initiate central nervous system infection in mice. Infect. Immun. 2001, 69, 1344–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drevets, D.A.; Leenen, P.J.; Greenfield, R.A. Invasion of the central nervous system by intracellular bacteria. Clin. Microbiol. Rev. 2004, 17, 323–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, W.K.; Harboe, Z.B.; Roed, C.; Monrad, J.B.; Lindelof, M.; Larsen, V.A.; Kondziella, D. Early trigeminal nerve involvement in Listeria monocytogenes rhombencephalitis: Case series and systematic review. J. Neurol. 2017, 264, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Pägelow, D.; Chhatbar, C.; Beineke, A.; Liu, X.; Nerlich, A.; van Vorst, K.; Rohde, M.; Kalinke, U.; Förster, R.; Halle, S.; et al. The olfactory epithelium as a port of entry in neonatal neurolisteriosis. Nat. Commun. 2018, 9, 4269. [Google Scholar] [CrossRef]
- Disson, O.; Lecuit, M. Targeting of the central nervous system by Listeria monocytogenes. Virulence 2012, 3, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Gründler, T.; Quednau, N.; Stump, C.; Orian-Rousseau, V.; Ishikawa, H.; Wolburg, H.; Schroten, H.; Tenenbaum, T.; Schwerk, C. The surface proteins InlA and InlB are interdependently required for polar basolateral invasion by Listeria monocytogenes in a human model of the blood-cerebrospinal fluid barrier. Microbes Infect. 2013, 15, 291–301. [Google Scholar] [CrossRef]
- Drevets, D.A.; Sawyer, R.T.; Potter, T.A.; Campbell, P.A. Listeria monocytogenes infects human endothelial cells by two distinct mechanisms. Infect. Immun. 1995, 63, 4268–4276. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, B.; Raffelsbauer, D.; Kuhn, M.; Goetz, M.; Hom, S.; Goebel, W. InlA- but not InlB-mediated internalization of Listeria monocytogenes by non-phagocytic mammalian cells needs the support of other internalins. Mol. Microbiol. 2002, 43, 557–570. [Google Scholar] [CrossRef]
- Dinner, S.; Kaltschmidt, J.; Stump-Guthier, C.; Hetjens, S.; Ishikawa, H.; Tenenbaum, T.; Schroten, H.; Schwerk, C. Mitogen-activated protein kinases are required for effective infection of human choroid plexus epithelial cells by Listeria monocytogenes. Microbes Infect. 2017, 19, 18–33. [Google Scholar] [CrossRef]
- Ghosh, P.; Halvorsen, E.M.; Ammendolia, D.A.; Mor-Vaknin, N.; O’Riordan, M.X.D.; Brumell, J.H.; Markovitz, D.M.; Higgins, D.E. Invasion of the brain by Listeria monocytogenes is mediated by InlF and host cell Vimentin. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antal, E.-A.; Løberg, E.M.; Bracht, P.; Melby, K.K.; Maehlen, J. Evidence for intraaxonal spread of listeria monocytogenes from the periphery to the central nervous system. Brain Pathol. 2001, 11, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Oevermann, A.; Zurbriggen, A.; Vandevelde, M. Rhombencephalitis caused by Listeria monocytogenes in humans and ruminants: A zoonosis on the rise? Interdiscip. Perspect. Infect. Dis. 2010, 2010, 632513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camejo, A.; Carvalho, F.; Reis, O.; Leitão, E.; Sousa, S.; Cabanes, D. The arsenal of virulence factors deployed by Listeria monocytogenes to promote its cell infection cycle. Virulence 2011, 2, 379–394. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.H.; Orsi, R.H.; Nightingale, K.K.; Wiedmann, M. Listeria monocytogenes internalins are highly diverse and evolved by recombination and positive selection. Infect. Genet. Evol. 2006, 6, 378–389. [Google Scholar] [CrossRef]
- Bierne, H.; Sabet, C.; Personnic, N.; Cossart, P. Internalins: A complex family of leucine-rich repeat-containing proteins in Listeria monocytogenes. Microbes Infect. 2007, 9, 1156–1166. [Google Scholar] [CrossRef]
- Harter, E.; Lassnig, C.; Wagner, E.M.; Zaiser, A.; Wagner, M.; Rychli, K. The novel internalins InlP1 and InlP4 and the internalin-Like Protein InlP3 Enhance the Pathogenicity of Listeria monocytogenes. Front. Microbiol. 2019, 10, 1644. [Google Scholar] [CrossRef] [Green Version]
- Kobe, B.; Kajava, A.V. The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 2001, 11, 725–732. [Google Scholar] [CrossRef]
- Gaillard, J.-L.; Berche, P.; Frehel, C.; Gouln, E.; Cossart, P. Entry of L. monocytogenes into cells is mediated by internalin, a repeat protein reminiscent of surface antigens from gram-positive cocci. Cell 1991, 65, 1127–1141. [Google Scholar] [CrossRef]
- Lingnau, A.; Domann, E.; Hudel, M.; Bock, M.; Nichterlein, T.; Wehland, J.; Chakraborty, T. Expression of the Listeria monocytogenes EGD inlA and inlB genes, whose products mediate bacterial entry into tissue culture cell lines, by PrfA-dependent and -independent mechanisms. Infect. Immun. 1995, 63, 3896–3903. [Google Scholar] [CrossRef] [Green Version]
- Parida, S.K.; Domann, E.; Rohde, M.; Müller, S.; Darji, A.; Hain, T.; Wehland, J.; Chakraborty, T. Internalin B is essential for adhesion and mediates the invasion of Listeria monocytogenes into human endothelial cells. Mol. Microbiol. 1998, 28, 81–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajabian, T.; Gavicherla, B.; Heisig, M.; Müller-Altrock, S.; Goebel, W.; Gray-Owen, S.D.; Ireton, K. The bacterial virulence factor InlC perturbs apical cell junctions and promotes cell-to-cell spread of Listeria. Nat. Cell Biol. 2009, 11, 1212–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dortet, L.; Mostowy, S.; Samba-Louaka, A.; Gouin, E.; Nahori, M.A.; Wiemer, E.A.; Dussurget, O.; Cossart, P. Recruitment of the major vault protein by InlK: A Listeria monocytogenes strategy to avoid autophagy. PLoS Pathog. 2011, 7, e1002168. [Google Scholar] [CrossRef]
- Faralla, C.; Bastounis, E.E.; Ortega, F.E.; Light, S.H.; Rizzuto, G.; Gao, L.; Marciano, D.K.; Nocadello, S.; Anderson, W.F.; Robbins, J.R.; et al. Listeria monocytogenes InlP interacts with afadin and facilitates basement membrane crossing. PLoS Pathog. 2018, 14, e1007094. [Google Scholar] [CrossRef] [Green Version]
- Lindén, S.K.; Bierne, H.; Sabet, C.; Png, C.W.; Florin, T.H.; McGuckin, M.A.; Cossart, P. Listeria monocytogenes internalins bind to the human intestinal mucin MUC2. Arch. Microbiol. 2008, 190, 101–104. [Google Scholar] [CrossRef]
- Phelps, C.C.; Vadia, S.; Arnett, E.; Tan, Y.; Zhang, X.; Pathak-Sharma, S.; Gavrilin, M.A.; Seveau, S. Relative roles of listeriolysin O, InlA, and InlB in Listeria monocytogenes uptake by host cells. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [Green Version]
- Dramsi, S.; Kocks, C.; Forestier, C.; Cossart, P. Internalin-mediated invasion of epithelial cells by Listeria monocytogenes is regulated by the bacterial growth state, temperature and the pleiotropic activator prfA. Mol. Microbiol. 1993, 9, 931–941. [Google Scholar] [CrossRef]
- Lecuit, M.; Vandormael-Pournin, S.; Lefort, J.; Huerre, M.; Gounon, P.; Dupuy, C.; Babinet, C.; Cossart, P. A transgenic model for listeriosis: Role of internalin in crossing the intestinal barrier. Science 2001, 292, 1722–1725. [Google Scholar] [CrossRef] [Green Version]
- Jacquet, C.; Doumith, M.; Gordon, J.I.; Martin, P.M.; Cossart, P.; Lecuit, M. A molecular marker for evaluating the pathogenic potential of foodborne Listeria monocytogenes. J. Infect. Dis 2004, 189, 2094–2100. [Google Scholar] [CrossRef] [Green Version]
- Prats, N.; Briones, V.; Blanco, M.M.; Altimira, J.; Ramos, J.A.; Domínguez, L.; Marco, A. Choroiditis and meningitis in experimental murine infection with Listeria monocytogenes. Eur. J. Clin. Microbiol. Infect. Dis 1992, 11, 744–747. [Google Scholar] [CrossRef]
- Schwerk, C.; Tenenbaum, T.; Kim, K.S.; Schroten, H. The choroid plexus-a multi-role player during infectious diseases of the CNS. Front. Cell Neurosci. 2015, 9, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizarro-Cerdá, J.; Kühbacher, A.; Cossart, P. Entry of Listeria monocytogenes in mammalian epithelial cells: An updated view. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Bonazzi, M.; Veiga, E.; Pizarro-Cerdá, J.; Cossart, P. Successive post-translational modifications of E-cadherin are required for InlA-mediated internalization of Listeria monocytogenes. Cell Microbiol. 2008, 10, 2208–2222. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Krause, G.; Scheffner, M.; Zechner, D.; Leddy, H.E.; Behrens, J.; Sommer, T.; Birchmeier, W. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol. 2002, 4, 222–231. [Google Scholar] [CrossRef]
- McLachlan, R.W.; Kraemer, A.; Helwani, F.M.; Kovacs, E.M.; Yap, A.S. E-cadherin adhesion activates c-Src signaling at cell-cell contacts. Mol. Biol. Cell 2007, 18, 3214–3223. [Google Scholar] [CrossRef] [Green Version]
- Sousa, S.; Cabanes, D.; Bougnères, L.; Lecuit, M.; Sansonetti, P.; Tran-Van-Nhieu, G.; Cossart, P. Src, cortactin and Arp2/3 complex are required for E-cadherin-mediated internalization of Listeria into cells. Cell Microbiol. 2007, 9, 2629–2643. [Google Scholar] [CrossRef]
- Veiga, E.; Guttman, J.A.; Bonazzi, M.; Boucrot, E.; Toledo-Arana, A.; Lin, A.E.; Enninga, J.; Pizarro-Cerdá, J.; Finlay, B.B.; Kirchhausen, T.; et al. Invasive and adherent bacterial pathogens co-opt host clathrin for infection. Cell Host Microbe 2007, 2, 340–351. [Google Scholar] [CrossRef] [Green Version]
- Bonazzi, M.; Vasudevan, L.; Mallet, A.; Sachse, M.; Sartori, A.; Prevost, M.-C.; Roberts, A.; Taner, S.B.; Wilbur, J.D.; Brodsky, F.M.; et al. Clathrin phosphorylation is required for actin recruitment at sites of bacterial adhesion and internalization. J. Cell Biol. 2011, 195, 525–536. [Google Scholar] [CrossRef]
- Lecuit, M.; Hurme, R.; Pizarro-Cerdá, J.; Ohayon, H.; Geiger, B.; Cossart, P. A role for alpha-and beta-catenins in bacterial uptake. Proc. Natl. Acad. Sci. USA 2000, 97, 10008–10013. [Google Scholar] [CrossRef] [Green Version]
- Lecuit, M.; Dramsi, S.; Gottardi, C.; Fedor-Chaiken, M.; Gumbiner, B.; Cossart, P. A single amino acid in E-cadherin responsible for host specificity towards the human pathogen Listeria monocytogenes. Embo J. 1999, 18, 3956–3963. [Google Scholar] [CrossRef] [Green Version]
- Dramsi, S.; Biswas, I.; Maguin, E.; Braun, L.; Mastroeni, P.; Cossart, P. Entry of Listeria monocytogenes into hepatocytes requires expression of inIB, a surface protein of the internalin multigene family. Mol. Microbiol. 1995, 16, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Braun, L.; Ghebrehiwet, B.; Cossart, P. gC1q-R/p32, a C1q-binding protein, is a receptor for the InlB invasion protein of Listeria monocytogenes. Embo J. 2000, 19, 1458–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Naujokas, M.; Park, M.; Ireton, K. InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell 2000, 103, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Jonquieres, R.; Pizarro-Cerda, J.; Cossart, P. Synergy between the N- and C-terminal domains of InlB for efficient invasion of non-phagocytic cells by Listeria monocytogenes. Mol. Microbiol. 2001, 42, 955–965. [Google Scholar] [CrossRef] [Green Version]
- Braun, L.; Ohayon, H.; Cossart, P. The InIB protein of Listeria monocytogenes is sufficient to promote entry into mammalian cells. Mol. Microbiol. 1998, 27, 1077–1087. [Google Scholar] [CrossRef]
- Braun, L.; Nato, F.; Payrastre, B.; Mazié, J.C.; Cossart, P. The 213-amino-acid leucine-rich repeat region of the listeria monocytogenes InlB protein is sufficient for entry into mammalian cells, stimulation of PI 3-kinase and membrane ruffling. Mol. Microbiol. 1999, 34, 10–23. [Google Scholar] [CrossRef]
- Hermiston, M.L.; Gordon, J.I. In vivo analysis of cadherin function in the mouse intestinal epithelium: Essential roles in adhesion, maintenance of differentiation, and regulation of programmed cell death. J. Cell Biol. 1995, 129, 489–506. [Google Scholar] [CrossRef]
- Greiffenberg, L.; Goebel, W.; Kim, K.S.; Daniels, J.; Kuhn, M. Interaction of Listeria monocytogenes with human brain microvascular endothelial cells: An electron microscopic study. Infect. Immun. 2000, 68, 3275–3279. [Google Scholar] [CrossRef] [Green Version]
- Hertzig, T.; Weber, M.; Greiffenberg, L.; Holthausen, B.S.; Goebel, W.; Kim, K.S.; Kuhn, M. Antibodies present in normal human serum inhibit invasion of human brain microvascular endothelial cells by Listeria monocytogenes. Infect. Immun. 2003, 71, 95–100. [Google Scholar] [CrossRef] [Green Version]
- Veiga, E.; Cossart, P. Listeria hijacks the clathrin-dependent endocytic machinery to invade mammalian cells. Nat. Cell Biol. 2005, 7, 894–900. [Google Scholar] [CrossRef]
- Ferraris, D.M.; Gherardi, E.; Di, Y.; Heinz, D.W.; Niemann, H.H. Ligand-mediated dimerization of the Met receptor tyrosine kinase by the bacterial invasion protein InlB. J. Mol. Biol. 2010, 395, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Ireton, K.; Payrastre, B.; Chap, H.; Ogawa, W.; Sakaue, H.; Kasuga, M.; Cossart, P. A role for phosphoinositide 3-kinase in bacterial invasion. Science 1996, 274, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Bierne, H.; Gouin, E.; Roux, P.; Caroni, P.; Yin, H.L.; Cossart, P. A role for cofilin and LIM kinase in Listeria-induced phagocytosis. J. Cell Biol. 2001, 155, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seveau, S.; Bierne, H.; Giroux, S.; Prévost, M.-C.; Cossart, P. Role of lipid rafts in E-cadherin—And HGF-R/Met—Mediated entry of Listeria monocytogenes into host cells. J. Cell Biol. 2004, 166, 743–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierne, H.; Miki, H.; Innocenti, M.; Scita, G.; Gertler, F.B.; Takenawa, T.; Cossart, P. WASP-related proteins, Abi1 and Ena/VASP are required for Listeria invasion induced by the Met receptor. J. Cell Sci. 2005, 118, 1537–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosse, T.; Ehinger, J.; Czuchra, A.; Benesch, S.; Steffen, A.; Wu, X.; Schloen, K.; Niemann, H.H.; Scita, G.; Stradal, T.E.; et al. Cdc42 and phosphoinositide 3-kinase drive Rac-mediated actin polymerization downstream of c-Met in distinct and common pathways. Mol. Cell Biol. 2007, 27, 6615–6628. [Google Scholar] [CrossRef] [Green Version]
- Jiwani, S.; Wang, Y.; Dowd, G.C.; Gianfelice, A.; Pichestapong, P.; Gavicherla, B.; Vanbennekom, N.; Ireton, K. Identification of components of the host type IA phosphoinositide 3-kinase pathway that promote internalization of Listeria monocytogenes. Infect. Immun. 2012, 80, 1252–1266. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.; Matzke, A.; Niemann, H.H.; Schwerk, C.; Tenenbaum, T.; Orian-Rousseau, V. Involvement of CD44v6 in InlB-dependent Listeria invasion. Mol. Microbiol. 2009, 72, 1196–1207. [Google Scholar] [CrossRef]
- Orian-Rousseau, V.; Chen, L.; Sleeman, J.P.; Herrlich, P.; Ponta, H. CD44 is required for two consecutive steps in HGF/c-Met signaling. Genes Dev. 2002, 16, 3074–3086. [Google Scholar] [CrossRef] [Green Version]
- Marino, M.; Braun, L.; Cossart, P.; Ghosh, P. Structure of the lnlB leucine-rich repeats, a domain that triggers host cell invasion by the bacterial pathogen L. monocytogenes. Mol. Cell 1999, 4, 1063–1072. [Google Scholar] [CrossRef]
- Dortet, L.; Veiga, E.; Bonazzi, M.; Cossart, P. CD44-independent activation of the Met signaling pathway by HGF and InlB. Microbes Infect. 2010, 12, 919–927. [Google Scholar] [CrossRef]
- Khelef, N.; Lecuit, M.; Bierne, H.; Cossart, P. Species specificity of the Listeria monocytogenes InlB protein. Cell Microbiol. 2006, 8, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Dramsi, S.; Dehoux, P.; Lebrun, M.; Goossens, P.L.; Cossart, P. Identification of four new members of the internalin multigene family of Listeria monocytogenes EGD. Infect. Immun. 1997, 65, 1615–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riento, K.; Ridley, A.J. Rocks: Multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, M.; Higgins, D.E. Inhibition of ROCK activity allows InlF-mediated invasion and increased virulence of Listeria monocytogenes. Mol. Microbiol. 2008, 68, 749–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noma, K.; Oyama, N.; Liao, J.K. Physiological role of ROCKs in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2006, 290, C661–C668. [Google Scholar] [CrossRef]
- Bastounis, E.E.; Yeh, Y.T.; Theriot, J.A. Matrix stiffness modulates infection of endothelial cells by Listeria monocytogenes via expression of cell surface vimentin. Mol. Biol. Cell 2018, 29, 1571–1589. [Google Scholar] [CrossRef]
- Hahmann, C.; Schroeter, T. Rho-kinase inhibitors as therapeutics: From pan inhibition to isoform selectivity. Cell Mol. Life Sci. 2010, 67, 171–177. [Google Scholar] [CrossRef]
- Sabet, C.; Lecuit, M.; Cabanes, D.; Cossart, P.; Bierne, H. LPXTG protein InlJ, a newly identified internalin involved in Listeria monocytogenes virulence. Infect. Immun. 2005, 73, 6912–6922. [Google Scholar] [CrossRef] [Green Version]
- Bublitz, M.; Holland, C.; Sabet, C.; Reichelt, J.; Cossart, P.; Heinz, D.W.; Bierne, H.; Schubert, W.D. Crystal structure and standardized geometric analysis of InlJ, a listerial virulence factor and leucine-rich repeat protein with a novel cysteine ladder. J. Mol. Biol. 2008, 378, 87–96. [Google Scholar] [CrossRef]
- Sabet, C.; Toledo-Arana, A.; Personnic, N.; Lecuit, M.; Dubrac, S.; Poupel, O.; Gouin, E.; Nahori, M.A.; Cossart, P.; Bierne, H. The Listeria monocytogenes virulence factor InlJ is specifically expressed in vivo and behaves as an adhesin. Infect. Immun. 2008, 76, 1368–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balandyte, L.; Brodard, I.; Frey, J.; Oevermann, A.; Abril, C. Ruminant rhombencephalitis-associated Listeria monocytogenes alleles linked to a multilocus variable-number tandem-repeat analysis complex. Appl. Environ. Microbiol. 2011, 77, 8325–8335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hain, T.; Steinweg, C.; Chakraborty, T. Comparative and functional genomics of Listeria spp. J. Biotechnol. 2006, 126, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Autret, N.; Dubail, I.; Trieu-Cuot, P.; Berche, P.; Charbit, A. Identification of new genes involved in the virulence of Listeria monocytogenes by signature-tagged transposon mutagenesis. Infect. Immun. 2001, 69, 2054–2065. [Google Scholar] [CrossRef] [Green Version]
- Popowska, M.; Krawczyk-Balska, A.; Ostrowski, R.; Desvaux, M. InlL from Listeria monocytogenes Is involved in biofilm formation and adhesion to mucin. Front. Microbiol. 2017, 8, 660. [Google Scholar] [CrossRef] [Green Version]
- Cabanes, D.; Dussurget, O.; Dehoux, P.; Cossart, P. Auto, a surface associated autolysin of Listeria monocytogenes required for entry into eukaryotic cells and virulence. Mol. Microbiol. 2004, 51, 1601–1614. [Google Scholar] [CrossRef]
- Popowska, M. Analysis of the peptidoglycan hydrolases of Listeria monocytogenes: Multiple enzymes with multiple functions. Pol. J. Microbiol. 2004, 53 (Suppl.), 29–34. [Google Scholar]
- Bublitz, M.; Polle, L.; Holland, C.; Heinz, D.W.; Nimtz, M.; Schubert, W.D. Structural basis for autoinhibition and activation of Auto, a virulence-associated peptidoglycan hydrolase of Listeria monocytogenes. Mol. Microbiol. 2009, 71, 1509–1522. [Google Scholar] [CrossRef]
- Wang, L.; Lin, M. Identification of IspC, an 86-kilodalton protein target of humoral immune response to infection with Listeria monocytogenes serotype 4b, as a novel surface autolysin. J. Bacteriol. 2007, 189, 2046–2054. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Lin, M. A novel cell wall-anchored peptidoglycan hydrolase (autolysin), IspC, essential for Listeria monocytogenes virulence: Genetic and proteomic analysis. Microbiology 2008, 154, 1900–1913. [Google Scholar] [CrossRef] [Green Version]
- Hamon, M.A.; Ribet, D.; Stavru, F.; Cossart, P. Listeriolysin O: The Swiss army knife of Listeria. Trends Microbiol. 2012, 20, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Osborne, S.E.; Brumell, J.H. Listeriolysin O: From bazooka to Swiss army knife. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christie, M.P.; Johnstone, B.A.; Tweten, R.K.; Parker, M.W.; Morton, C.J. Cholesterol-dependent cytolysins: From water-soluble state to membrane pore. Biophys. Rev. 2018, 10, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Schuerch, D.W.; Wilson-Kubalek, E.M.; Tweten, R.K. Molecular basis of listeriolysin O pH dependence. Proc. Natl. Acad. Sci. USA 2005, 102, 12537–12542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Nguyen, B.N.; Mitchell, G.; Margolis, S.R.; Ma, D.; Portnoy, D.A. The Listeriolysin O PEST-like Sequence Co-opts AP-2-Mediated Endocytosis to Prevent Plasma Membrane Damage during Listeria Infection. Cell Host Microbe 2018, 23, 786–795. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, J.L.; Berche, P.; Sansonetti, P. Transposon mutagenesis as a tool to study the role of hemolysin in the virulence of Listeria monocytogenes. Infect. Immun. 1986, 52, 50–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossart, P.; Vicente, M.F.; Mengaud, J.; Baquero, F.; Perez-Diaz, J.C.; Berche, P. Listeriolysin O is essential for virulence of Listeria monocytogenes: Direct evidence obtained by gene complementation. Infect. Immun. 1989, 57, 3629–3636. [Google Scholar] [CrossRef] [Green Version]
- Gekara, N.O.; Westphal, K.; Ma, B.; Rohde, M.; Groebe, L.; Weiss, S. The multiple mechanisms of Ca2+ signalling by listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Cell Microbiol. 2007, 9, 2008–2021. [Google Scholar] [CrossRef]
- Stavru, F.; Bouillaud, F.; Sartori, A.; Ricquier, D.; Cossart, P. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc. Natl. Acad. Sci. USA 2011, 108, 3612–3617. [Google Scholar] [CrossRef] [Green Version]
- Vadia, S.; Arnett, E.; Haghighat, A.C.; Wilson-Kubalek, E.M.; Tweten, R.K.; Seveau, S. The pore-forming toxin listeriolysin O mediates a novel entry pathway of L. monocytogenes into human hepatocytes. PLoS Pathog. 2011, 7, e1002356. [Google Scholar] [CrossRef] [Green Version]
- Vadia, S.; Seveau, S. Fluxes of Ca2+ and K+ are required for the listeriolysin O-dependent internalization pathway of Listeria monocytogenes. Infect. Immun. 2014, 82, 1084–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Bae, D.; Wang, C. Listeriolysin O mediates cytotoxicity against human brain microvascular endothelial cells. FEMS Microbiol. Lett. 2015, 362, fnv084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, J.G.T.; Vadia, S.; Pathak-Sharma, S.; McLaughlin, E.; Zhang, X.; Swanson, J.; Seveau, S. Host cell perforation by listeriolysin O (LLO) activates a Ca (2+)-dependent cPKC/Rac1/Arp2/3 signaling pathway that promotes Listeria monocytogenes internalization independently of membrane resealing. Mol. Biol. Cell 2018, 29, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Eisenreich, W.; Dandekar, T.; Heesemann, J.; Goebel, W. Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat. Rev. Microbiol 2010, 8, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Le Guennec, L.; Coureuil, M.; Nassif, X.; Bourdoulous, S. Strategies used by bacterial pathogens to cross the blood–brain barrier. Cell Microbiol. 2020, 22, e13132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herold, R.; Schroten, H.; Schwerk, C. Virulence factors of meningitis-causing bacteria: Enabling brain entry across the blood-brain barrier. Int. J. Mol. Sci. 2019, 20, 5393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guldimann, C.; Lejeune, B.; Hofer, S.; Leib, S.L.; Frey, J.; Zurbriggen, A.; Seuberlich, T.; Oevermann, A. Ruminant organotypic brain-slice cultures as a model for the investigation of CNS listeriosis. Int. J. Exp. Pathol. 2012, 93, 259–268. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banović, F.; Schroten, H.; Schwerk, C. Potential Roles and Functions of Listerial Virulence Factors during Brain Entry. Toxins 2020, 12, 297. https://doi.org/10.3390/toxins12050297
Banović F, Schroten H, Schwerk C. Potential Roles and Functions of Listerial Virulence Factors during Brain Entry. Toxins. 2020; 12(5):297. https://doi.org/10.3390/toxins12050297
Chicago/Turabian StyleBanović, Franjo, Horst Schroten, and Christian Schwerk. 2020. "Potential Roles and Functions of Listerial Virulence Factors during Brain Entry" Toxins 12, no. 5: 297. https://doi.org/10.3390/toxins12050297
APA StyleBanović, F., Schroten, H., & Schwerk, C. (2020). Potential Roles and Functions of Listerial Virulence Factors during Brain Entry. Toxins, 12(5), 297. https://doi.org/10.3390/toxins12050297