Repurposing Cancer Drugs Batimastat and Marimastat to Inhibit the Activity of a Group I Metalloprotease from the Venom of the Western Diamondback Rattlesnake, Crotalus atrox

 , ,
, , 
Abstract
:1. Introduction
2. Results
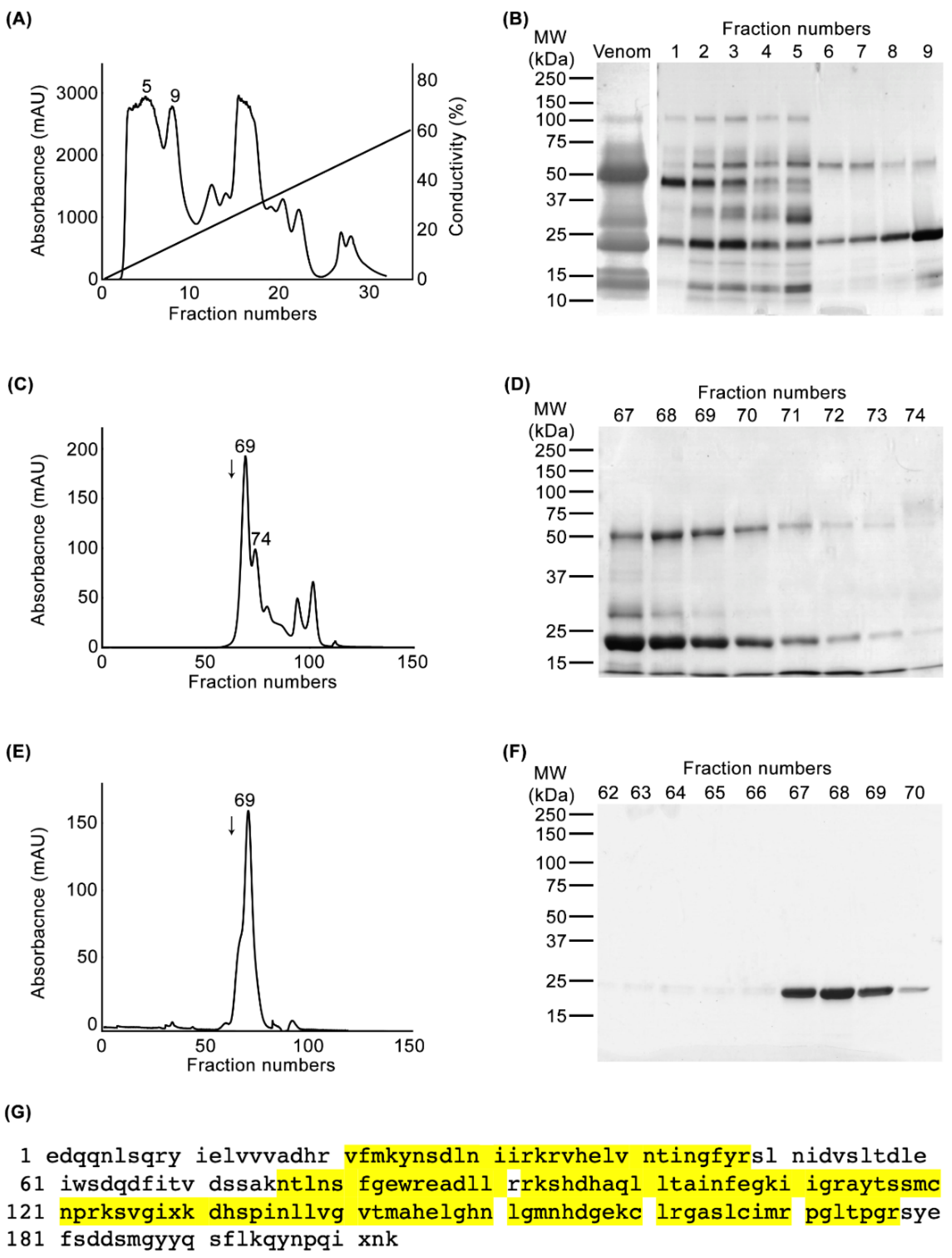
2.1. Purification and Identification of CAMP-2
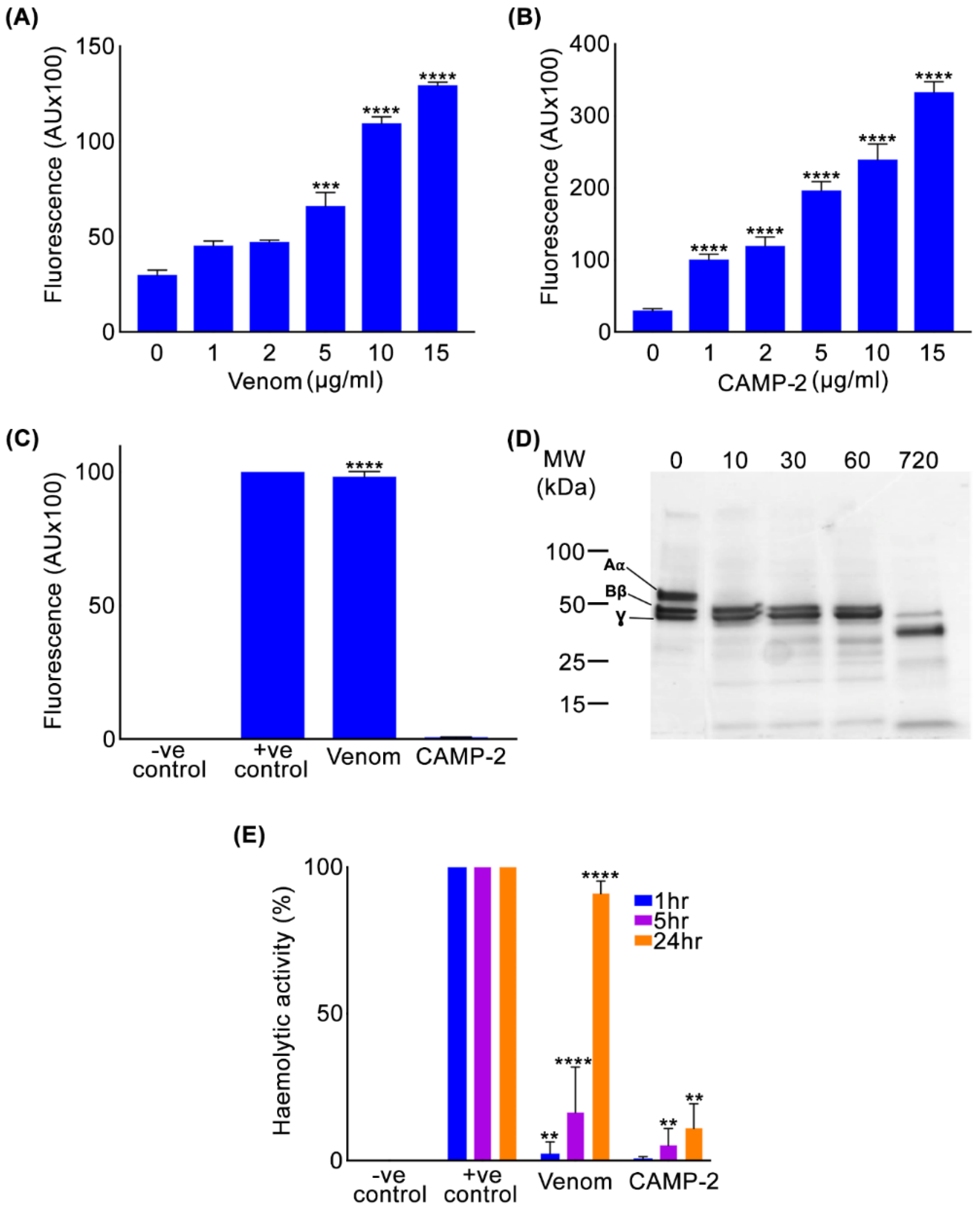
2.2. CAMP-2 Exerts Collagenolytic, Fibrinogenolytic and Haemolytic Activities
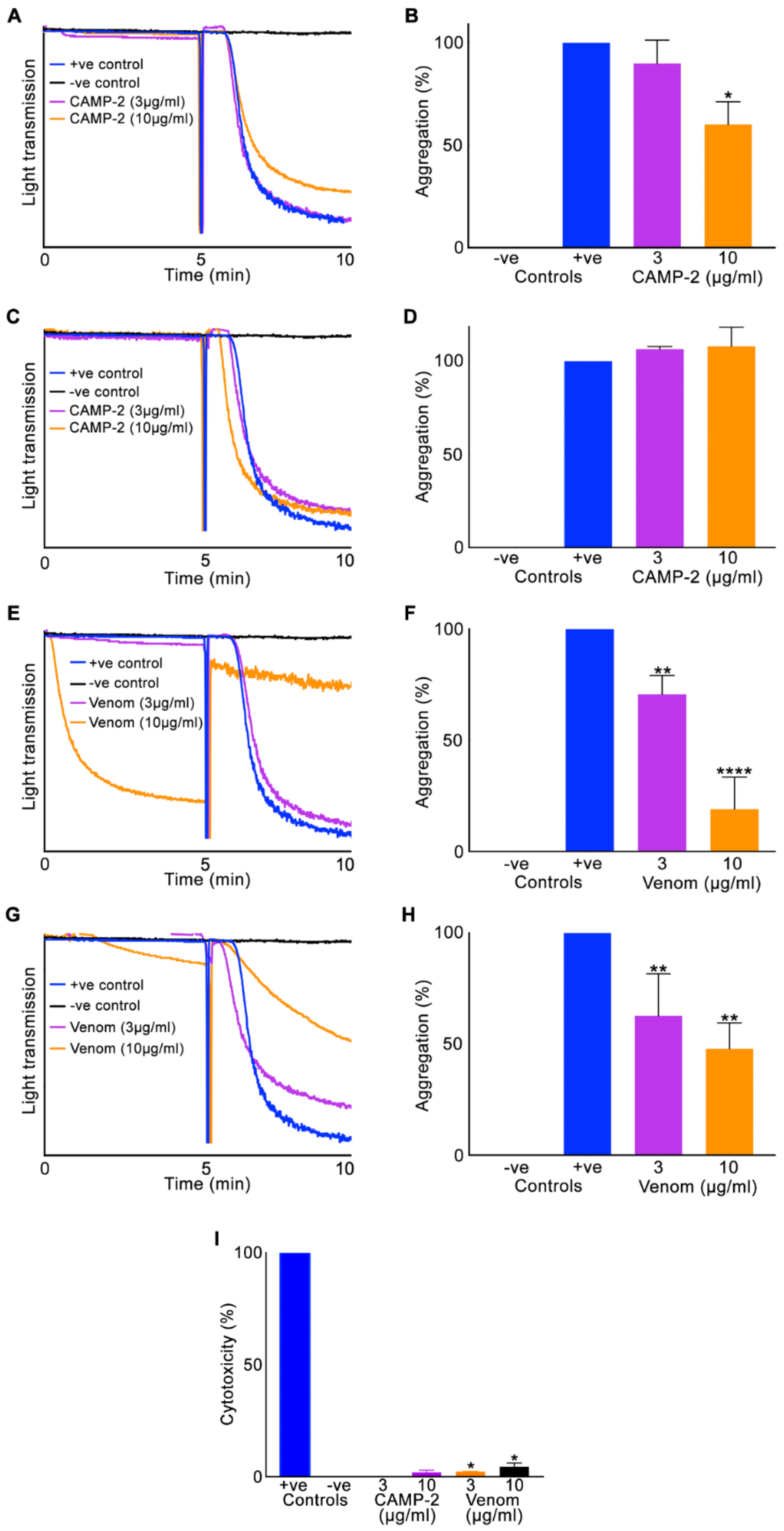
2.3. CAMP-2 Inhibits Human Platelet Aggregation
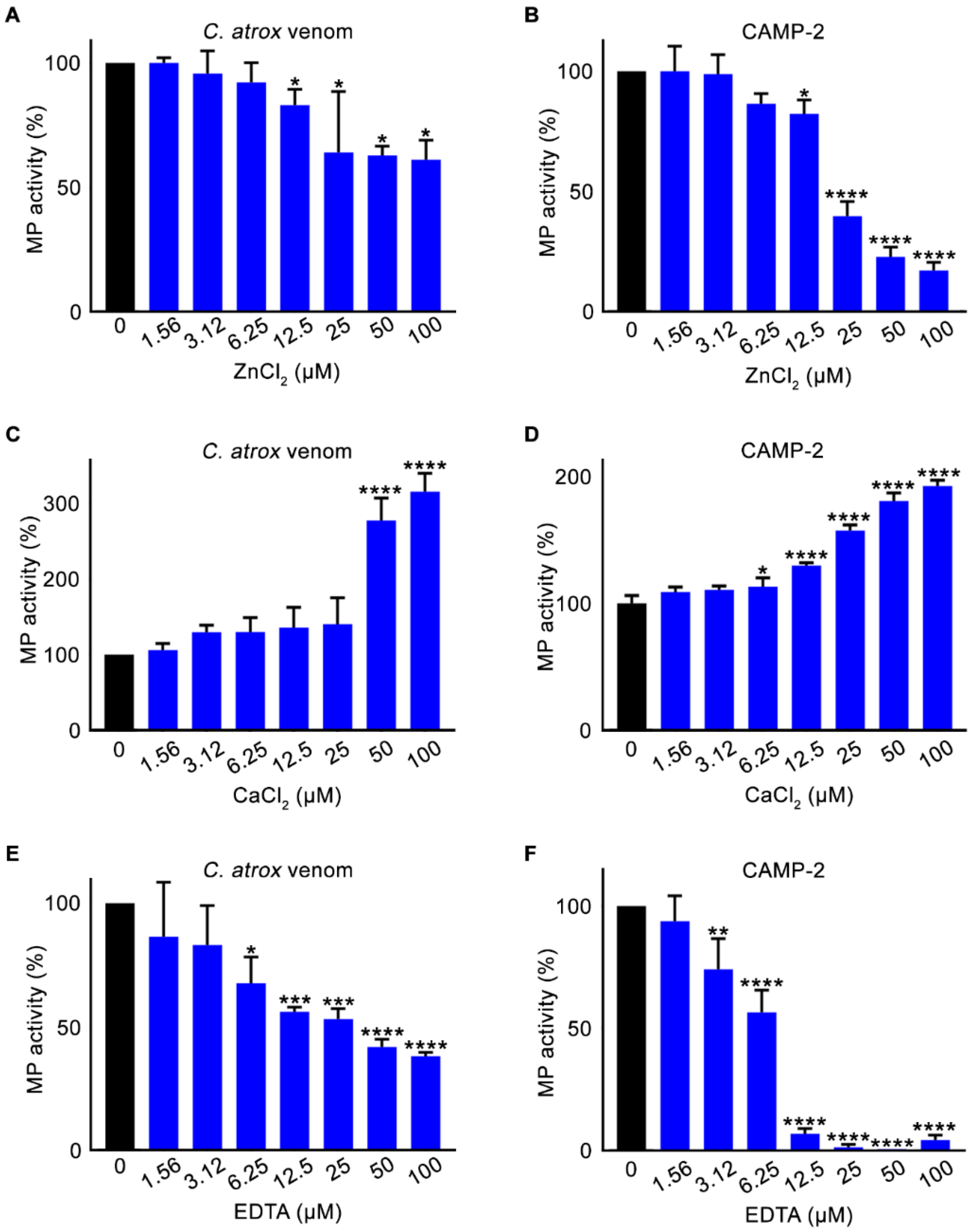
2.4. Chlorides and a Metal Chelator Affect Metalloprotease Activity of CAMP-2
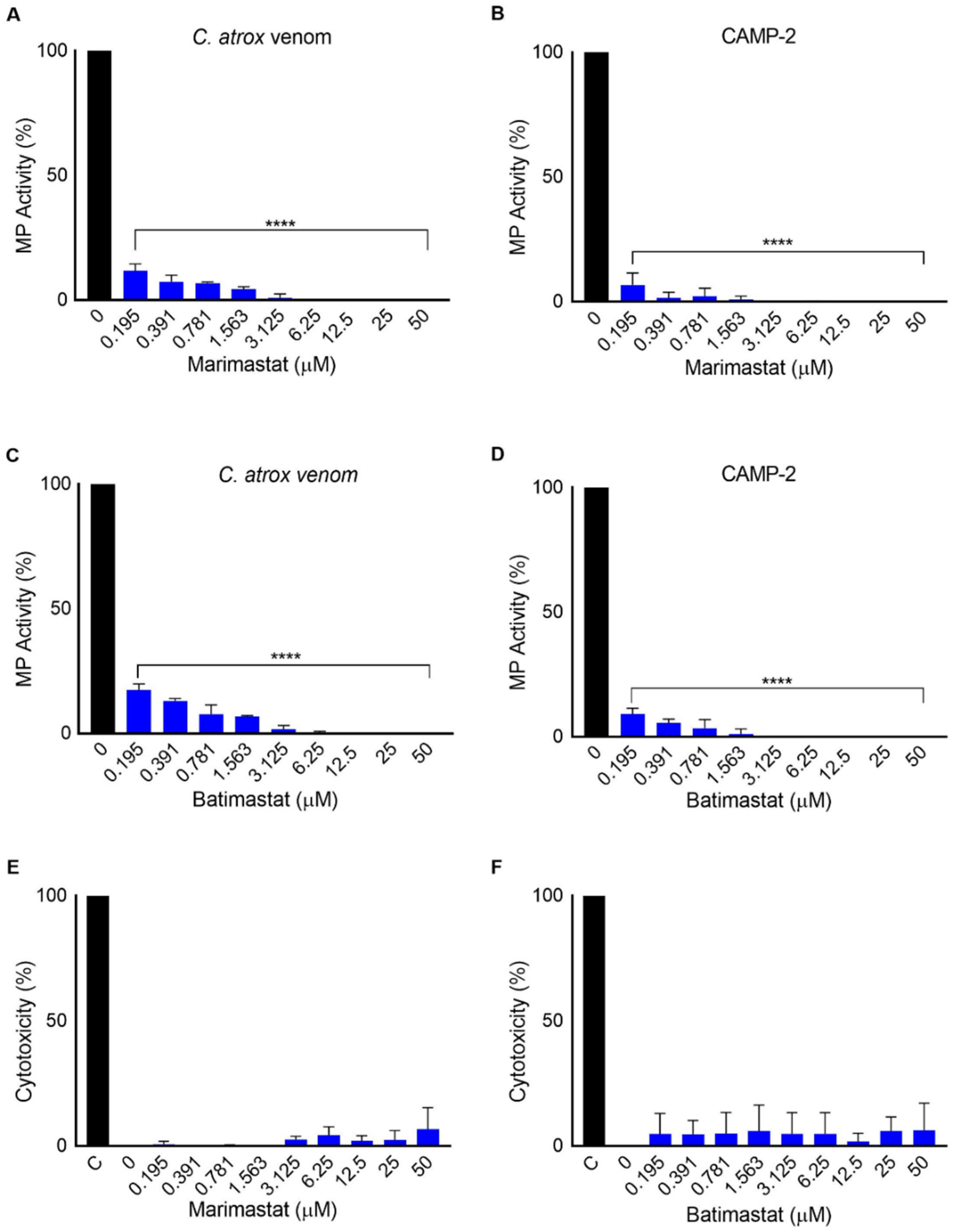
2.5. Marimastat and Batimastat Inhibit the Activity of CAMP-2
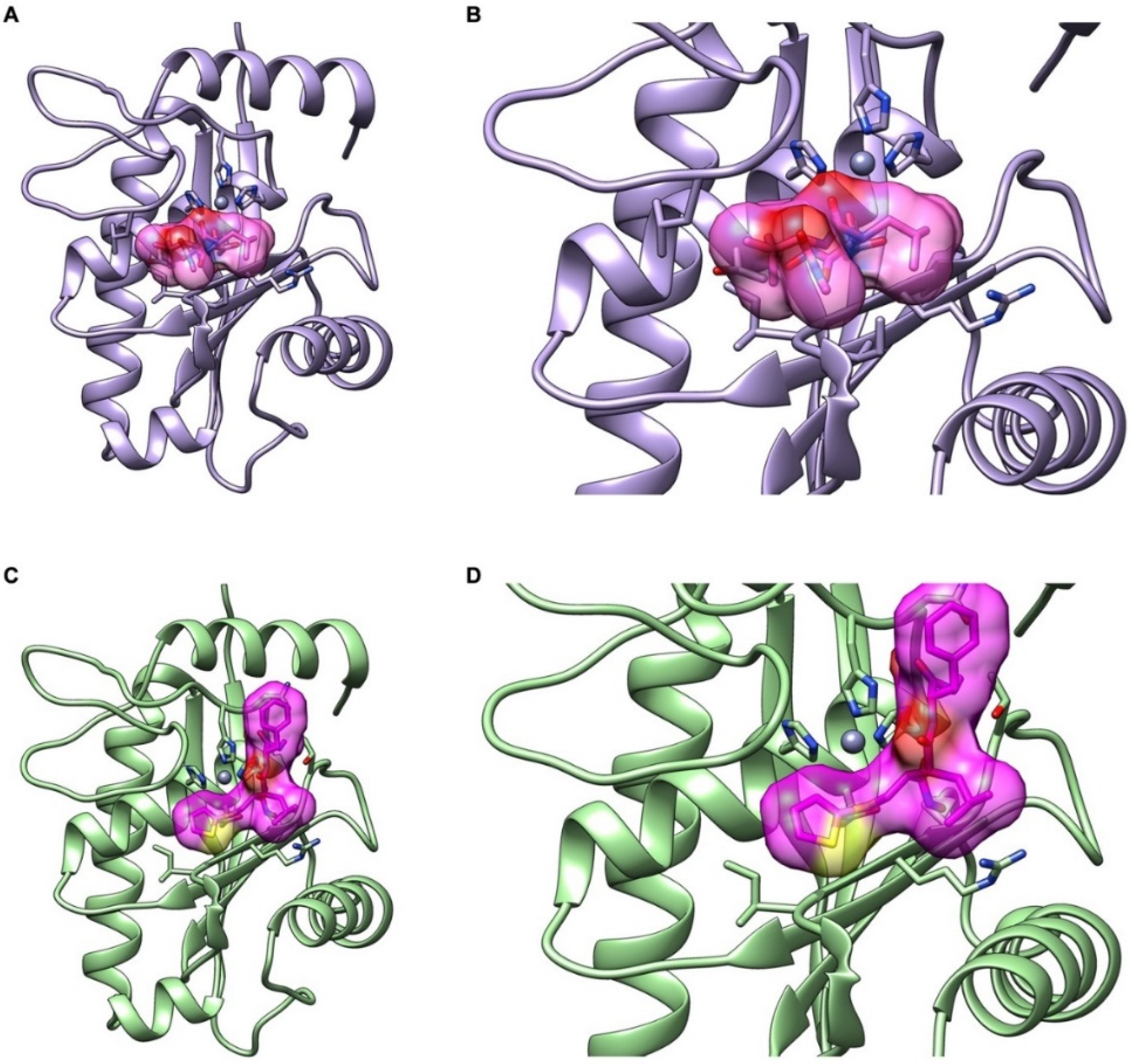
2.6. Interactions of CAMP-2 with Batimastat and Marimastat
3. Discussion
4. Materials and Methods
4.1. Protein Purification
4.2. Mass Spectrometry Analysis
4.3. Fluorogenic Assays
4.4. Fibrinogenolytic Assay
4.5. Human Blood Collection, Platelet Preparation and Aggregation Assay
4.6. Haemolytic Assay
4.7. LDH Cytotoxicity Assay
4.8. Structure Modelling and Molecular Docking
4.9. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Gutierrez, J.M.; Calvete, J.J.; Habib, A.G.; Harrison, R.A.; Williams, D.J.; Warrell, D.A. Snakebite envenoming. Nat. Rev. Dis. Primers 2017, 3, 17079. [Google Scholar] [CrossRef]
- Chippaux, J.P. Snakebite envenomation turns again into a neglected tropical disease! J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 38. [Google Scholar] [CrossRef]
- Kasturiratne, A.; Wickremasinghe, A.R.; de Silva, N.; Gunawardena, N.K.; Pathmeswaran, A.; Premaratna, R.; Savioli, L.; Lalloo, D.G.; de Silva, H.J. The global burden of snakebite: A literature analysis and modelling based on regional estimates of envenoming and deaths. PLoS Med. 2008, 5, e218. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.; Gutierrez, J.M.; Harrison, R.; Warrell, D.A.; White, J.; Winkel, K.D.; Gopalakrishnakone, P. The Global Snake Bite Initiative: An antidote for snake bite. Lancet 2010, 375, 89–91. [Google Scholar] [CrossRef]
- Williams, H.F.; Layfield, H.J.; Vallance, T.; Patel, K.; Bicknell, A.B.; Trim, S.A.; Vaiyapuri, S. The Urgent Need to Develop Novel Strategies for the Diagnosis and Treatment of Snakebites. Toxins (Basel) 2019, 11, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunagar, K.; Undheim, E.A.; Scheib, H.; Gren, E.C.; Cochran, C.; Person, C.E.; Koludarov, I.; Kelln, W.; Hayes, W.K.; King, G.F.; et al. Intraspecific venom variation in the medically significant Southern Pacific Rattlesnake (Crotalus oreganus helleri): Biodiscovery, clinical and evolutionary implications. J. Proteom. 2014, 99, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G.; Winkel, K.D.; Wickramaratna, J.C.; Hodgson, W.C.; Wüster, W. Effectiveness of Snake Antivenom: Species and Regional Venom Variation and Its Clinical Impact. J. Toxicol. Toxin Rev. 2003, 22, 23–34. [Google Scholar] [CrossRef]
- Pla, D.; Sanz, L.; Sasa, M.; Acevedo, M.E.; Dwyer, Q.; Durban, J.; Perez, A.; Rodriguez, Y.; Lomonte, B.; Calvete, J.J. Proteomic analysis of venom variability and ontogeny across the arboreal palm-pitvipers (genus Bothriechis). J. Proteom. 2017, 152, 1–12. [Google Scholar] [CrossRef]
- Modahl, C.M.; Mukherjee, A.K.; Mackessy, S.P. An analysis of venom ontogeny and prey-specific toxicity in the Monocled Cobra (Naja kaouthia). Toxicon 2016, 119, 8–20. [Google Scholar] [CrossRef]
- Harrison, R.A.; Hargreaves, A.; Wagstaff, S.C.; Faragher, B.; Lalloo, D.G. Snake envenoming: A disease of poverty. PLoS Negl. Trop. Dis. 2009, 3, e569. [Google Scholar] [CrossRef] [Green Version]
- Yanez-Arenas, C.; Peterson, A.T.; Mokondoko, P.; Rojas-Soto, O.; Martinez-Meyer, E. The use of ecological niche modeling to infer potential risk areas of snakebite in the Mexican state of Veracruz. PLoS ONE 2014, 9, e100957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvete, J.J.; Fasoli, E.; Sanz, L.; Boschetti, E.; Righetti, P.G. Exploring the venom proteome of the western diamondback rattlesnake, Crotalus atrox, via snake venomics and combinatorial peptide ligand library approaches. J. Proteome Res. 2009, 8, 3055–3067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, J.M.; Rucavado, A. Snake venom metalloproteinases: Their role in the pathogenesis of local tissue damage. Biochimie 2000, 82, 841–850. [Google Scholar] [CrossRef]
- Williams, H.F.; Mellows, B.A.; Mitchell, R.; Sfyri, P.; Layfield, H.J.; Salamah, M.; Vaiyapuri, R.; Collins-Hooper, H.; Bicknell, A.B.; Matsakas, A.; et al. Mechanisms underpinning the permanent muscle damage induced by snake venom metalloprotease. PLoS Negl. Trop. Dis. 2019, 13, e0007041. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.W.; Serrano, S.M. Structural considerations of the snake venom metalloproteinases, key members of the M12 reprolysin family of metalloproteinases. Toxicon 2005, 45, 969–985. [Google Scholar] [CrossRef]
- Rasmussen, H.S.; McCann, P.P. Matrix metalloproteinase inhibition as a novel anticancer strategy: A review with special focus on batimastat and marimastat. Pharm. Ther. 1997, 75, 69–75. [Google Scholar] [CrossRef]
- Knudsen, C.; Laustsen, A.H. Recent Advances in Next Generation Snakebite Antivenoms. Trop. Med. Infect. Dis. 2018, 3, 42. [Google Scholar] [CrossRef] [Green Version]
- Markland, F.S., Jr.; Swenson, S. Snake venom metalloproteinases. Toxicon 2013, 62, 3–18. [Google Scholar] [CrossRef]
- Baker, B.J.; Wongvibulsin, S.; Nyborg, J.; Tu, A.T. Nucleotide sequence encoding the snake venom fibrinolytic enzyme atroxase obtained from a Crotalus atrox venom gland cDNA library. Arch. Biochem. Biophys. 1995, 317, 357–364. [Google Scholar] [CrossRef]
- Willis, T.W.; Tu, A.T. Purification and biochemical characterization of atroxase, a nonhemorrhagic fibrinolytic protease from western diamondback rattlesnake venom. Biochemistry 1988, 27, 4769–4777. [Google Scholar] [CrossRef]
- Tu, A.T.; Baker, B.; Wongvibulsin, S.; Willis, T. Biochemical characterization of atroxase and nucleotide sequence encoding the fibrinolytic enzyme. Toxicon 1996, 34, 1295–1300. [Google Scholar] [CrossRef]
- Baker, B.J.; Tu, A.T. Atroxase—A fibrinolytic enzyme isolated from the venom of western diamondback rattlesnake. Isolation, characterization and cloning. Adv. Exp. Med. Biol. 1996, 391, 203–211. [Google Scholar] [PubMed]
- Willis, T.W.; Tu, A.T.; Miller, C.W. Thrombolysis with a snake venom protease in a rat model of venous thrombosis. Thromb. Res. 1989, 53, 19–29. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinform. 2008, 24, 8.14.1–8.14.40. [Google Scholar] [CrossRef] [PubMed]
- Vaiyapuri, S.; Vaiyapuri, R.; Ashokan, R.; Ramasamy, K.; Nattamaisundar, K.; Jeyaraj, A.; Chandran, V.; Gajjeraman, P.; Baksh, M.F.; Gibbins, J.M.; et al. Snakebite and its socio-economic impact on the rural population of Tamil Nadu, India. PLoS ONE 2013, 8, e80090. [Google Scholar] [CrossRef] [Green Version]
- Williams, H.F.; Vaiyapuri, R.; Gajjeraman, P.; Hutchinson, G.; Gibbins, J.M.; Bicknell, A.B.; Vaiyapuri, S. Challenges in diagnosing and treating snakebites in a rural population of Tamil Nadu, India: The views of clinicians. Toxicon 2017, 130, 44–46. [Google Scholar] [CrossRef] [Green Version]
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures Into Future Successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [Green Version]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef]
- Vaiyapuri, S.; Hutchinson, E.G.; Ali, M.S.; Dannoura, A.; Stanley, R.G.; Harrison, R.A.; Bicknell, A.B.; Gibbins, J.M. Rhinocetin, a venom-derived integrin-specific antagonist inhibits collagen-induced platelet and endothelial cell functions. J. Biol. Chem. 2012, 287, 26235–26244. [Google Scholar] [CrossRef] [Green Version]
- Howes, J.M.; Theakston, R.D.; Laing, G.D. Neutralization of the haemorrhagic activities of viperine snake venoms and venom metalloproteinases using synthetic peptide inhibitors and chelators. Toxicon 2007, 49, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, H.S. Batimastat and Marimastat in Cancer. In Antiangiogenic Agents in Cancer Therapy; Teicher, B.A., Ed.; Humana Press: Totowa, NJ, USA, 1999; pp. 399–405. [Google Scholar]
- Rucavado, A.; Escalante, T.; Gutiérrez, J.M.A. Effect of the metalloproteinase inhibitor batimastat in the systemic toxicity induced by Bothrops asper snake venom: Understanding the role of metalloproteinases in envenomation. Toxicon 2004, 43, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Rucavado, A.; Escalante, T.; Franceschi, A.; Chaves, F.; León, G.; Cury, Y.; Ovadia, M.; Gutiérrez, J.M. Inhibition of local hemorrhage and dermonecrosis induced by Bothrops asper snake venom: Effectiveness of early in situ administration of the peptidomimetic metalloproteinase inhibitor batimastat and the chelating agent CaNa2EDTA. Am. J. Trop. Med. Hyg. 2000, 63, 313–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escalante, T.; Franceschi, A.; Rucavado, A.; Gutiérrez, J.M.A. Effectiveness of batimastat, a synthetic inhibitor of matrix metalloproteinases, in neutralizing local tissue damage induced by BaP1, a hemorrhagic metalloproteinase from the venom of the snake Bothrops asper. Biochem. Pharm. 2000, 60, 269–274. [Google Scholar] [CrossRef]
- Ainsworth, S.; Slagboom, J.; Alomran, N.; Pla, D.; Alhamdi, Y.; King, S.I.; Bolton, F.M.S.; Gutierrez, J.M.; Vonk, F.J.; Toh, C.H.; et al. The paraspecific neutralisation of snake venom induced coagulopathy by antivenoms. Commun. Biol. 2018, 1, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordeiro, F.A.; Coutinho, B.M.; Wiezel, G.A.; Bordon, K.F. Purification and enzymatic characterization of a novel metalloprotease from Lachesis muta rhombeata snake venom. J. Venom. Anim. Toxins Incl. Trop. Dis. 2018, 24, 32. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Ortiz, M.; Gomis-Ruth, F.X.; Huber, R.; Aviles, F.X. Inhibition of carboxypeptidase A by excess zinc: Analysis of the structural determinants by X-ray crystallography. Febs Press 1997, 400, 336–340. [Google Scholar] [CrossRef] [Green Version]
- Takeda, S. ADAM and ADAMTS Family Proteins and Snake, Venom Metalloproteinases: A Structural Overview. Toxins 2016, 8, 155. [Google Scholar] [CrossRef] [Green Version]
- Kini, R.M.; Koh, C.Y. Metalloproteases Affecting Blood Coagulation, Fibrinolysis and Platelet Aggregation from Snake Venoms: Definition and Nomenclature of Interaction Sites. Toxins (Basel) 2016, 8, 284. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Morita, T. Snake venom components affecting blood coagulation and the vascular system: Structural similarities and marked diversity. Curr. Pharm. Des. 2007, 13, 2872–2886. [Google Scholar] [CrossRef]
- Vaiyapuri, S.; Sage, T.; Rana, R.H.; Schenk, M.P.; Ali, M.S.; Unsworth, A.J.; Jones, C.I.; Stainer, A.R.; Kriek, N.; Moraes, L.A.; et al. EphB2 regulates contact-dependent and contact-independent signaling to control platelet function. Blood 2015, 125, 720–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Binding Energy (kcal/mol) | Ligand Efficiency (kcal/mol) | Inhibitory Constant (μM) | H Bond Interactions (D-H…A) | Distance (Å) |
|---|---|---|---|---|---|
| Batimastat | −5.59 | −0.17 | 79.37 | ALA 114 N-H…O HIS 154 N-H…O | 2.8 2.8 |
| Marimastat | −5.29 | −0.23 | 132.52 | N-H…O LYS 109 ILE 111 N-H…O GLY 112 N-H…O N-H…O(E2) GLU 145 O-H…O(E2) GLU 145 | 3.2 3.2 3.0 2.6 3.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Layfield, H.J.; Williams, H.F.; Ravishankar, D.; Mehmi, A.; Sonavane, M.; Salim, A.; Vaiyapuri, R.; Lakshminarayanan, K.; Vallance, T.M.; Bicknell, A.B.; et al. Repurposing Cancer Drugs Batimastat and Marimastat to Inhibit the Activity of a Group I Metalloprotease from the Venom of the Western Diamondback Rattlesnake, Crotalus atrox. Toxins 2020, 12, 309. https://doi.org/10.3390/toxins12050309
Layfield HJ, Williams HF, Ravishankar D, Mehmi A, Sonavane M, Salim A, Vaiyapuri R, Lakshminarayanan K, Vallance TM, Bicknell AB, et al. Repurposing Cancer Drugs Batimastat and Marimastat to Inhibit the Activity of a Group I Metalloprotease from the Venom of the Western Diamondback Rattlesnake, Crotalus atrox. Toxins. 2020; 12(5):309. https://doi.org/10.3390/toxins12050309
Chicago/Turabian StyleLayfield, Harry J., Harry F. Williams, Divyashree Ravishankar, Amita Mehmi, Medha Sonavane, Anika Salim, Rajendran Vaiyapuri, Karthik Lakshminarayanan, Thomas M. Vallance, Andrew B. Bicknell, and et al. 2020. "Repurposing Cancer Drugs Batimastat and Marimastat to Inhibit the Activity of a Group I Metalloprotease from the Venom of the Western Diamondback Rattlesnake, Crotalus atrox" Toxins 12, no. 5: 309. https://doi.org/10.3390/toxins12050309
APA StyleLayfield, H. J., Williams, H. F., Ravishankar, D., Mehmi, A., Sonavane, M., Salim, A., Vaiyapuri, R., Lakshminarayanan, K., Vallance, T. M., Bicknell, A. B., Trim, S. A., Patel, K., & Vaiyapuri, S. (2020). Repurposing Cancer Drugs Batimastat and Marimastat to Inhibit the Activity of a Group I Metalloprotease from the Venom of the Western Diamondback Rattlesnake, Crotalus atrox. Toxins, 12(5), 309. https://doi.org/10.3390/toxins12050309