Multi-Stress Induction of the Mycobacterium tuberculosis MbcTA Bactericidal Toxin-Antitoxin System

 , and
, and 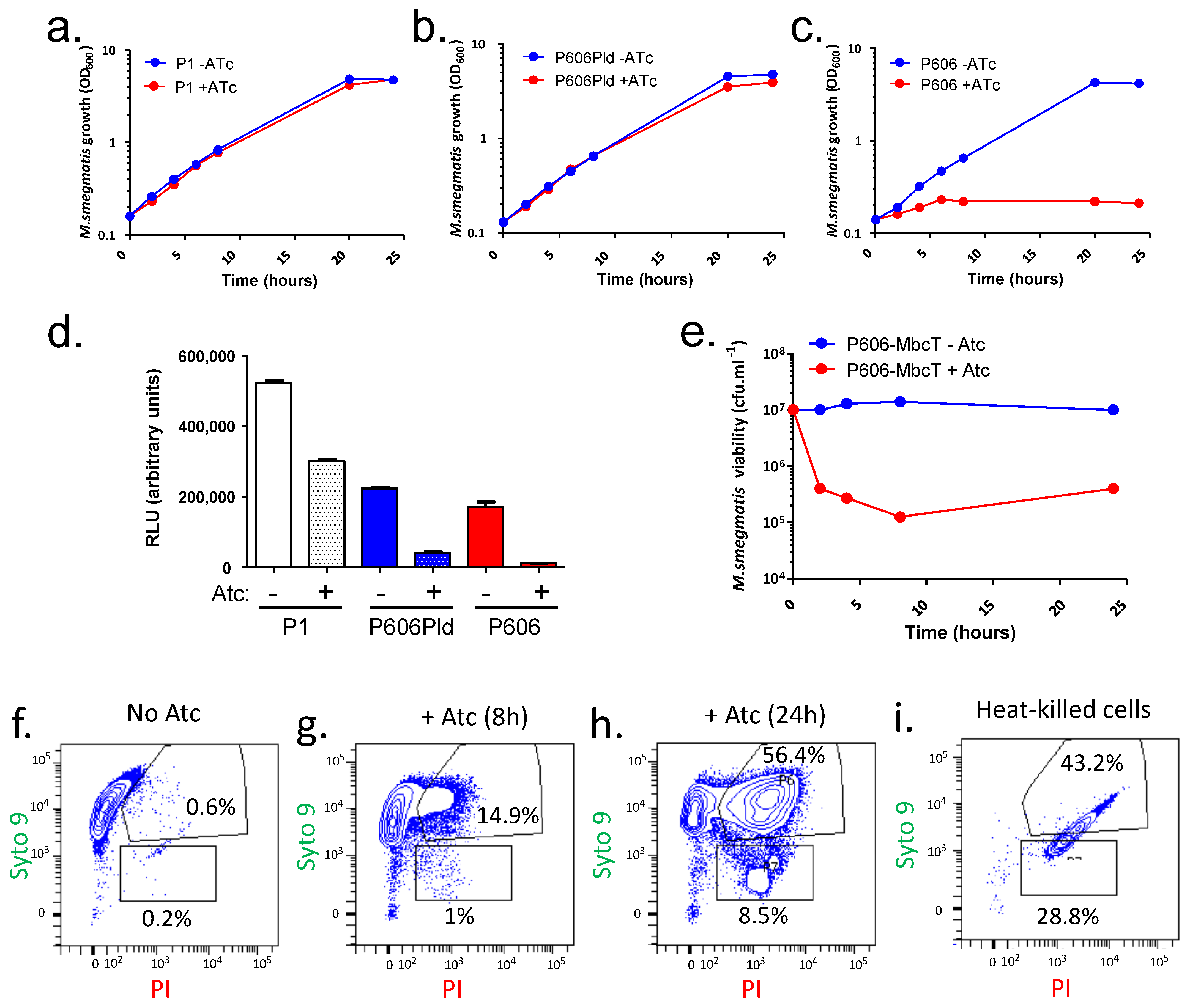{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. MbcT Toxin Depletes NAD+ and Is Bactericidal in Mycobacterium smegmatis
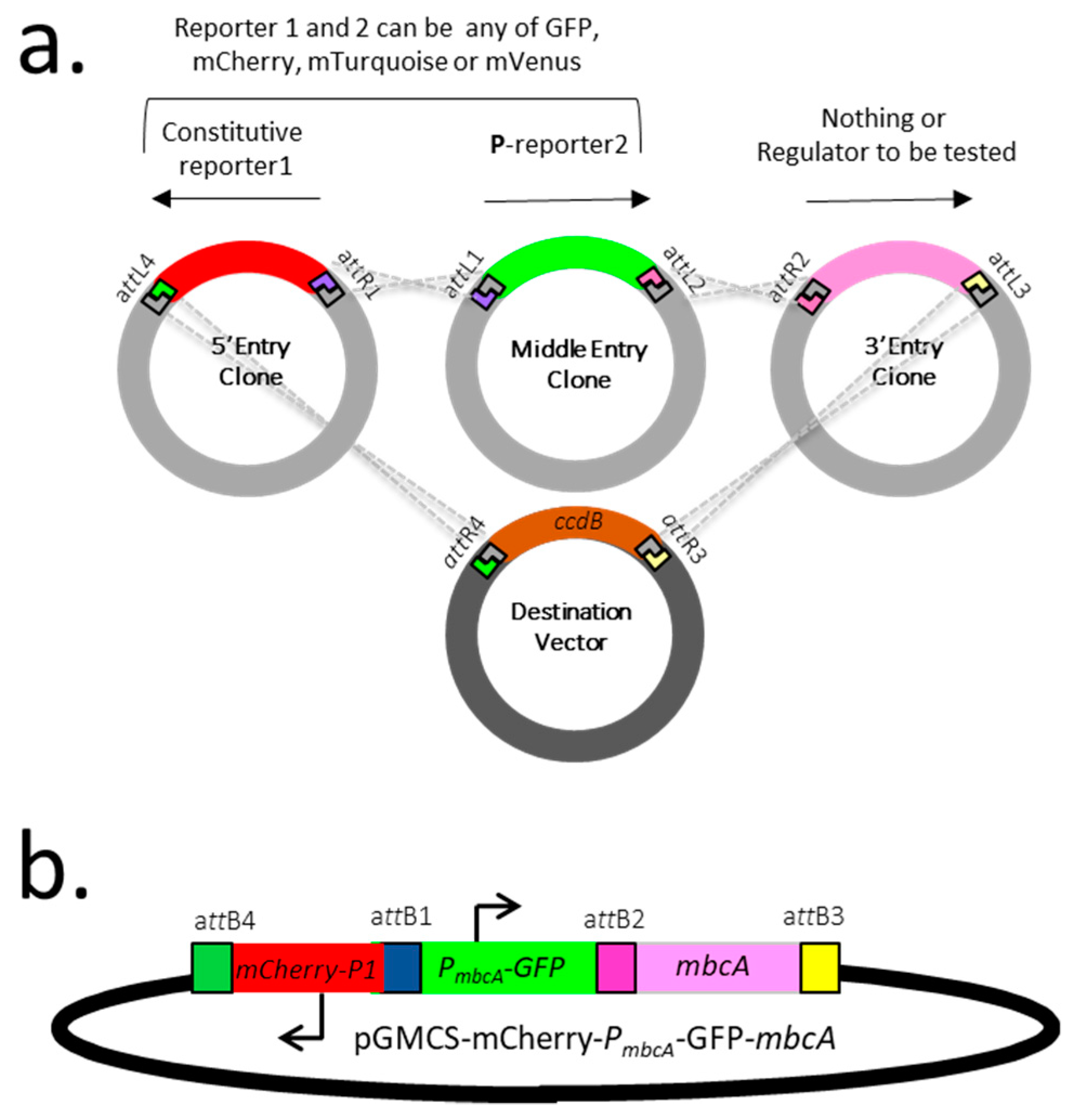
2.2. Construction of a Dual Fluorescent Reporter System to Assess Gene Expression in Mycobacteria
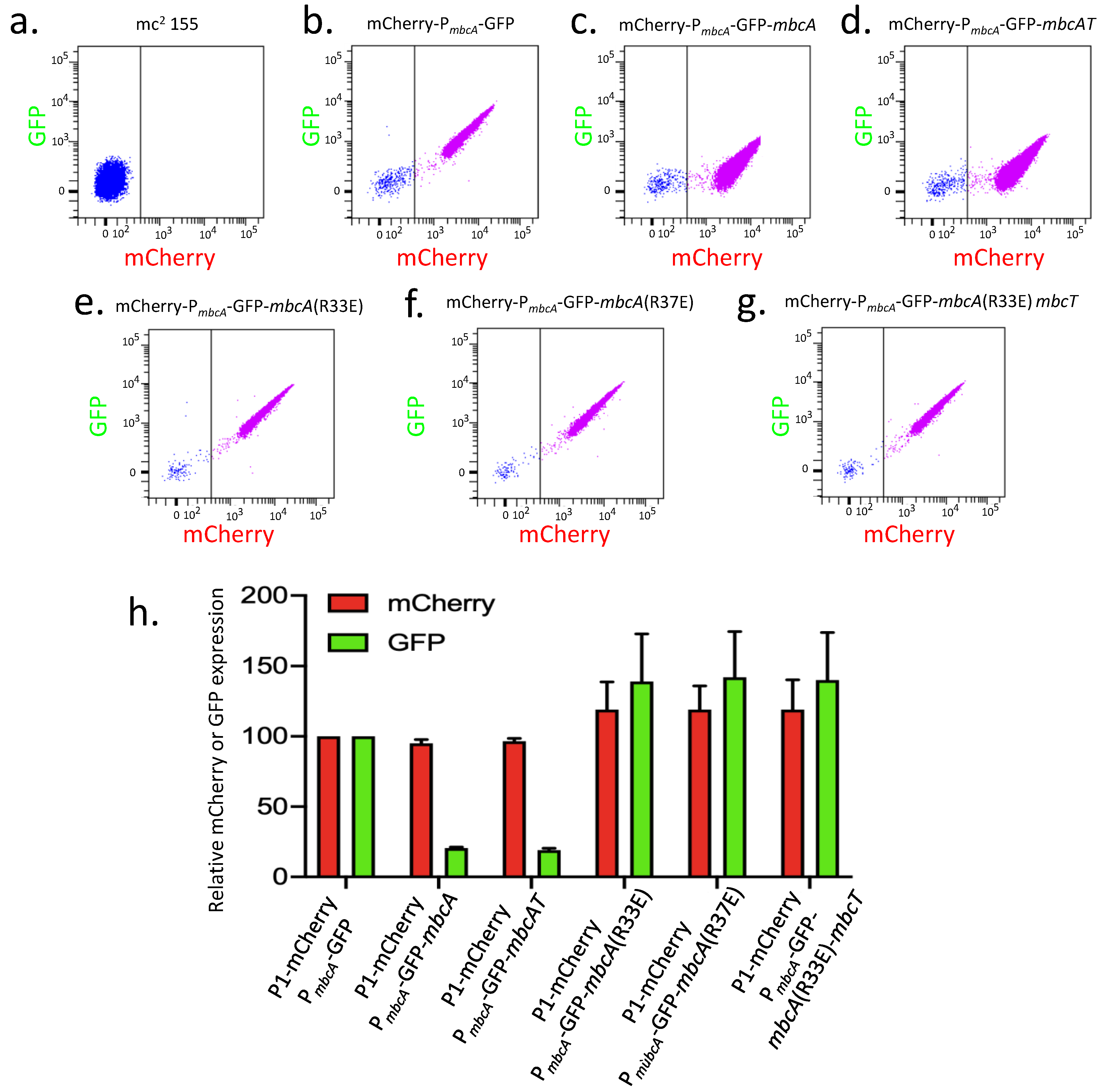
2.3. Use of a Dual Fluorescent Reporter System to Study the Expression of mbcAT
2.3.1. Auto-Repression of mbcAT
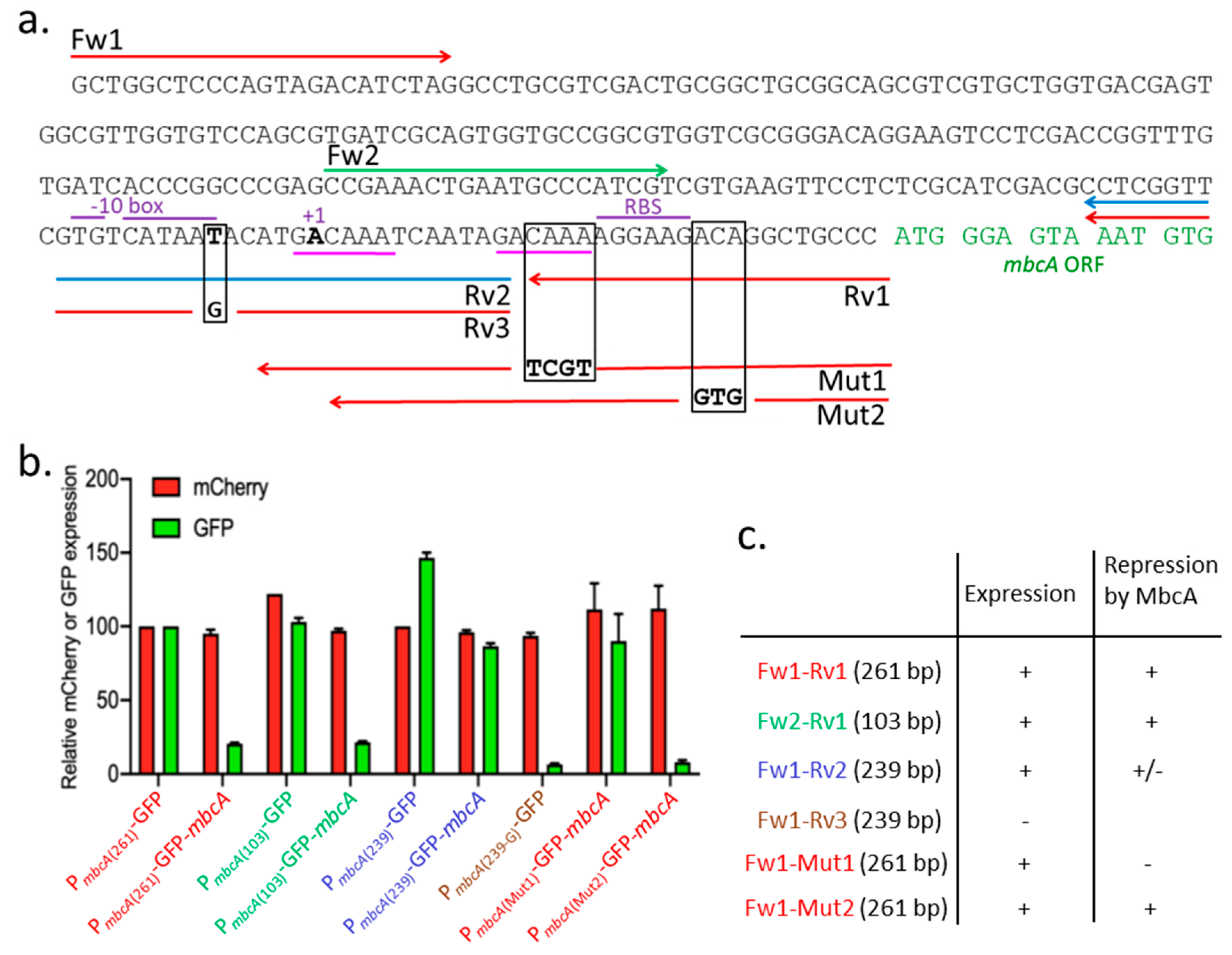
2.3.2. Genetic Dissection of the mbcAT Promoter/Operator Region
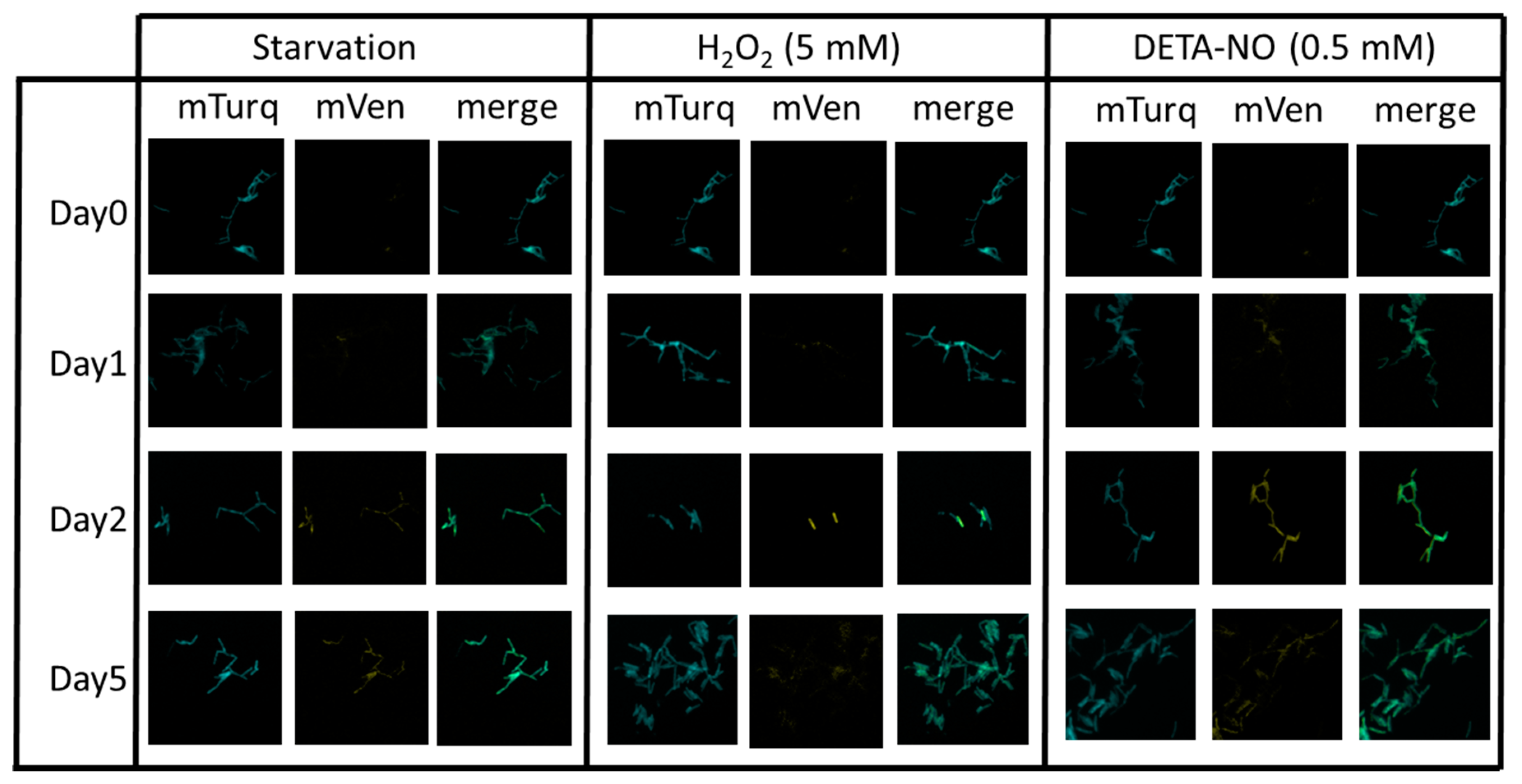
2.4. Multistress Induction of mbcAT Expression
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Cultures
4.2. Oligonucleotides and Plasmids Used in This Work
4.3. M. Smegmatis Viability Assays and NAD+ Measurement
4.4. Effect of Stress Conditions on PmbcA
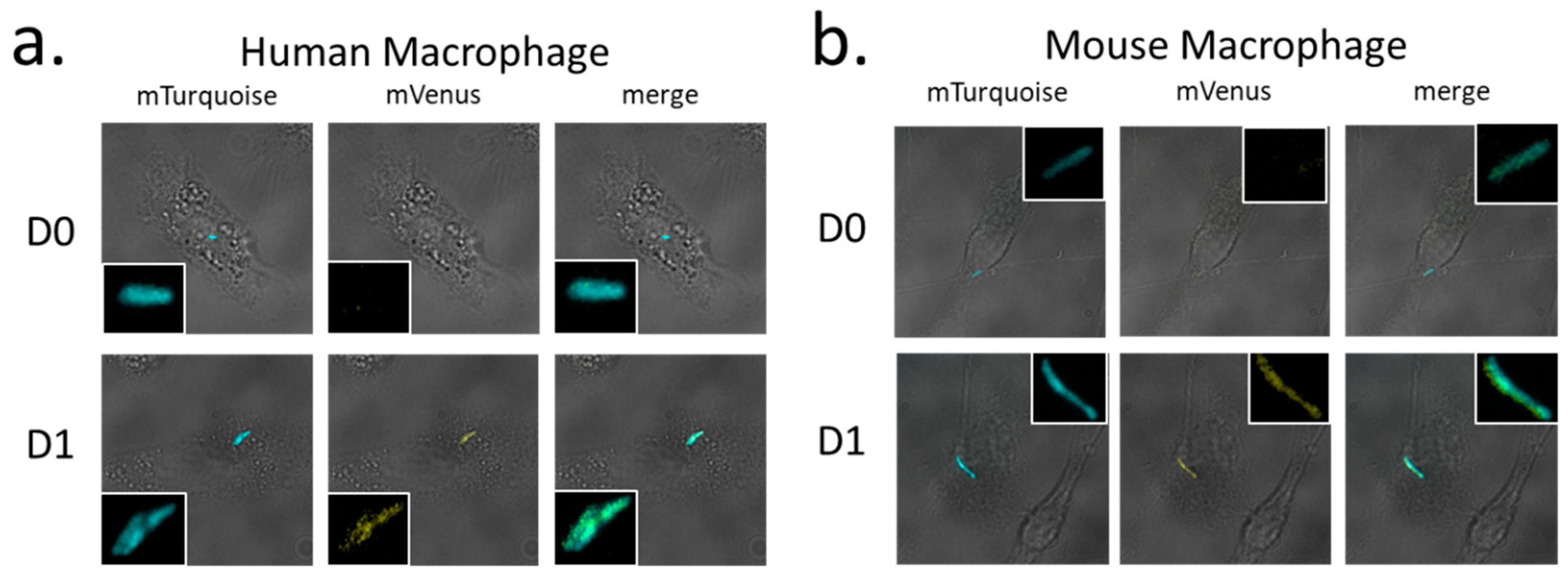
4.5. Human and Mice Macrophage Cultures and Infection
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Freire, D.M.; Gutierrez, C.; Garza-Garcia, A.; Grabowska, A.D.; Sala, A.J.; Ariyachaokun, K.; Panikova, T.; Beckham, K.S.H.; Colom, A.; Pogenberg, V.; et al. An NAD(+) Phosphorylase Toxin Triggers Mycobacterium tuberculosis Cell Death. Mol. Cell 2019, 73, 1282–1291.e8. [Google Scholar] [CrossRef] [Green Version]
- Harms, A.; Brodersen, D.E.; Mitarai, N.; Gerdes, K. Toxins, Targets, and Triggers: An Overview of Toxin-Antitoxin Biology. Mol. Cell 2018, 70, 768–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobato-Marquez, D.; Diaz-Orejas, R.; Garcia-Del Portillo, F. Toxin-antitoxins and bacterial virulence. FEMS Microbiol. Rev. 2016, 40, 592–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, R.; Peti, W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016, 12, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Deter, H.S.; Jensen, R.V.; Mather, W.H.; Butzin, N.C. Mechanisms for Differential Protein Production in Toxin-Antitoxin Systems. Toxins 2017, 9, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Wood, T.K. Toxin/Antitoxin System Paradigms: Toxins Bound to Antitoxins Are Not Likely Activated by Preferential Antitoxin Degradation. Adv. Biosyst. 2020, 4, e1900290. [Google Scholar] [CrossRef] [PubMed]
- Akarsu, H.; Bordes, P.; Mansour, M.; Bigot, D.J.; Genevaux, P.; Falquet, L. TASmania: A bacterial Toxin-Antitoxin Systems database. PLoS Comput. Biol. 2019, 15, e1006946. [Google Scholar] [CrossRef] [Green Version]
- Keren, I.; Minami, S.; Rubin, E.; Lewis, K. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. MBio 2011, 2, e00100–e00111. [Google Scholar] [CrossRef] [Green Version]
- Ramage, H.R.; Connolly, L.E.; Cox, J.S. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: Implications for pathogenesis, stress responses, and evolution. PLoS Genet. 2009, 5, e1000767. [Google Scholar] [CrossRef] [Green Version]
- Sala, A.; Bordes, P.; Genevaux, P. Multiple toxin-antitoxin systems in Mycobacterium tuberculosis. Toxins 2014, 6, 1002–1020. [Google Scholar] [CrossRef] [Green Version]
- Slayden, R.A.; Dawson, C.C.; Cummings, J.E. Toxin-antitoxin systems and regulatory mechanisms in Mycobacterium tuberculosis. Pathog. Dis. 2018, 76, fty039. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Pascopella, L.; Jacobs, W.R., Jr.; Hatfull, G.F. Site-specific integration of mycobacteriophage L5: Integration-proficient vectors for Mycobacterium smegmatis, Mycobacterium tuberculosis, and bacille Calmette-Guerin. Proc. Natl. Acad. Sci. USA 1991, 88, 3111–3115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrt, S.; Guo, X.V.; Hickey, C.M.; Ryou, M.; Monteleone, M.; Riley, L.W.; Schnappinger, D. Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res. 2005, 33, e21. [Google Scholar] [CrossRef] [PubMed]
- Schnappinger, D.; Ehrt, S. Regulated Expression Systems for Mycobacteria and Their Applications. Microbiol. Spectr. 2014, 2, MGM2-0018-2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, T.; Schubert, O.T.; Rose, G.; Arnvig, K.B.; Comas, I.; Aebersold, R.; Young, D.B. Genome-wide mapping of transcriptional start sites defines an extensive leaderless transcriptome in Mycobacterium tuberculosis. Cell Rep. 2013, 5, 1121–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levillain, F.; Poquet, Y.; Mallet, L.; Mazeres, S.; Marceau, M.; Brosch, R.; Bange, F.C.; Supply, P.; Magalon, A.; Neyrolles, O. Horizontal acquisition of a hypoxia-responsive molybdenum cofactor biosynthesis pathway contributed to Mycobacterium tuberculosis pathoadaptation. PLoS Pathog. 2017, 13, e1006752. [Google Scholar] [CrossRef] [Green Version]
- Thakur, Z.; Saini, V.; Arya, P.; Kumar, A.; Mehta, P.K. Computational insights into promoter architecture of toxin-antitoxin systems of Mycobacterium tuberculosis. Gene 2018, 641, 161–171. [Google Scholar] [CrossRef]
- Waagmeester, A.; Thompson, J.; Reyrat, J.M. Identifying sigma factors in Mycobacterium smegmatis by comparative genomic analysis. Trends Microbiol. 2005, 13, 505–509. [Google Scholar] [CrossRef]
- Garcia-Pino, A.; Balasubramanian, S.; Wyns, L.; Gazit, E.; De Greve, H.; Magnuson, R.D.; Charlier, D.; Van Nuland, N.A.; Loris, R. Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell 2010, 142, 101–111. [Google Scholar] [CrossRef]
- Song, S.; Wood, T.K. Post-segregational Killing and Phage Inhibition Are Not Mediated by Cell Death Through Toxin/Antitoxin Systems. Front. Microbiol. 2018, 9, 814. [Google Scholar] [CrossRef] [Green Version]
- Albrethsen, J.; Agner, J.; Piersma, S.R.; Hojrup, P.; Pham, T.V.; Weldingh, K.; Jimenez, C.R.; Andersen, P.; Rosenkrands, I. Proteomic profiling of Mycobacterium tuberculosis identifies nutrient-starvation-responsive toxin-antitoxin systems. Mol. Cell. Proteom. 2013, 12, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rustad, T.R.; Harrell, M.I.; Liao, R.; Sherman, D.R. The enduring hypoxic response of Mycobacterium tuberculosis. PLoS ONE 2008, 3, e1502. [Google Scholar] [CrossRef] [PubMed]
- Flannagan, R.S.; Cosio, G.; Grinstein, S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 2009, 7, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.G. Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunol. Rev. 2011, 240, 252–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsunawaki, S.; Nathan, C.F. Enzymatic basis of macrophage activation. Kinetic analysis of superoxide production in lysates of resident and activated mouse peritoneal macrophages and granulocytes. J. Biol. Chem. 1984, 259, 4305–4312. [Google Scholar] [PubMed]
- Dy, R.L.; Przybilski, R.; Semeijn, K.; Salmond, G.P.; Fineran, P.C. A widespread bacteriophage abortive infection system functions through a Type IV toxin-antitoxin mechanism. Nucleic Acids Res. 2014, 42, 4590–4605. [Google Scholar] [CrossRef]
- Pecota, D.C.; Wood, T.K. Exclusion of T4 phage by the hok/sok killer locus from plasmid R1. J. Bacteriol. 1996, 178, 2044–2050. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, S. Trouble is coming: Signaling pathways that regulate general stress responses in bacteria. J. Biol. Chem. 2019, 294, 11685–11700. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.Y.; Lee, B.J. Structure, Biology, and Therapeutic Application of Toxin-Antitoxin Systems in Pathogenic Bacteria. Toxins 2016, 8, 305. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.J.; Hergenrother, P.J. Artificial activation of toxin-antitoxin systems as an antibacterial strategy. Trends Microbiol. 2012, 20, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Helaine, S.; Cheverton, A.M.; Watson, K.G.; Faure, L.M.; Matthews, S.A.; Holden, D.W. Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science 2014, 343, 204–208. [Google Scholar] [CrossRef]
- Keren, I.; Kaldalu, N.; Spoering, A.; Wang, Y.; Lewis, K. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 2004, 230, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Ronneau, S.; Helaine, S. Clarifying the Link between Toxin-Antitoxin Modules and Bacterial Persistence. J. Mol. Biol. 2019, 431, 3462–3471. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wood, T.K. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 2011, 77, 5577–5583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraikin, N.; Goormaghtigh, F.; Van Melderen, L. Type II Toxin-Antitoxin Systems: Evolution and Revolutions. J. Bacteriol. 2020, 202, e00763-19. [Google Scholar] [CrossRef] [Green Version]
- DeJesus, M.A.; Gerrick, E.R.; Xu, W.; Park, S.W.; Long, J.E.; Boutte, C.C.; Rubin, E.J.; Schnappinger, D.; Ehrt, S.; Fortune, S.M.; et al. Comprehensive Essentiality Analysis of the Mycobacterium tuberculosis Genome via Saturating Transposon Mutagenesis. MBio 2017, 8, e02133-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; O’Brien, K.M.; Sharma, R.; Boshoff, H.I.; Rehren, G.; Chakraborty, S.; Wallach, J.B.; Monteleone, M.; Wilson, D.J.; Aldrich, C.C.; et al. A genetic strategy to identify targets for the development of drugs that prevent bacterial persistence. Proc. Natl. Acad Sci. USA 2013, 110, 19095–19100. [Google Scholar] [CrossRef] [Green Version]
- Rodionova, I.A.; Schuster, B.M.; Guinn, K.M.; Sorci, L.; Scott, D.A.; Li, X.; Kheterpal, I.; Shoen, C.; Cynamon, M.; Locher, C.; et al. Metabolic and bactericidal effects of targeted suppression of NadD and NadE enzymes in mycobacteria. MBio 2014, 5, e00747-13. [Google Scholar] [CrossRef] [Green Version]
- Vilcheze, C.; Weinrick, B.; Wong, K.W.; Chen, B.; Jacobs, W.R., Jr. NAD+ auxotrophy is bacteriocidal for the tubercle bacilli. Mol. Microbiol. 2010, 76, 365–377. [Google Scholar] [CrossRef] [Green Version]
- Troegeler, A.; Lastrucci, C.; Duval, C.; Tanne, A.; Cougoule, C.; Maridonneau-Parini, I.; Neyrolles, O.; Lugo-Villarino, G. An efficient siRNA-mediated gene silencing in primary human monocytes, dendritic cells and macrophages. Immunol. Cell Biol. 2014, 92, 699–708. [Google Scholar] [CrossRef]
- Benard, A.; Sakwa, I.; Schierloh, P.; Colom, A.; Mercier, I.; Tailleux, L.; Jouneau, L.; Boudinot, P.; Al-Saati, T.; Lang, R.; et al. B Cells Producing Type I IFN Modulate Macrophage Polarization in Tuberculosis. Am. J. Respir. Crit. Care Med. 2018, 197, 801–813. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ariyachaokun, K.; Grabowska, A.D.; Gutierrez, C.; Neyrolles, O. Multi-Stress Induction of the Mycobacterium tuberculosis MbcTA Bactericidal Toxin-Antitoxin System. Toxins 2020, 12, 329. https://doi.org/10.3390/toxins12050329
Ariyachaokun K, Grabowska AD, Gutierrez C, Neyrolles O. Multi-Stress Induction of the Mycobacterium tuberculosis MbcTA Bactericidal Toxin-Antitoxin System. Toxins. 2020; 12(5):329. https://doi.org/10.3390/toxins12050329
Chicago/Turabian StyleAriyachaokun, Kanchiyaphat, Anna D. Grabowska, Claude Gutierrez, and Olivier Neyrolles. 2020. "Multi-Stress Induction of the Mycobacterium tuberculosis MbcTA Bactericidal Toxin-Antitoxin System" Toxins 12, no. 5: 329. https://doi.org/10.3390/toxins12050329
APA StyleAriyachaokun, K., Grabowska, A. D., Gutierrez, C., & Neyrolles, O. (2020). Multi-Stress Induction of the Mycobacterium tuberculosis MbcTA Bactericidal Toxin-Antitoxin System. Toxins, 12(5), 329. https://doi.org/10.3390/toxins12050329