Mycotoxin Determination in Animal Feed: An LC-FLD Method for Simultaneous Quantification of Aflatoxins, Ochratoxins and Zearelanone in This Matrix


Abstract
:1. Introduction
2. Results
2.1. Sample Treatment
2.2. Chromatographic Conditions
2.3. Method Validation
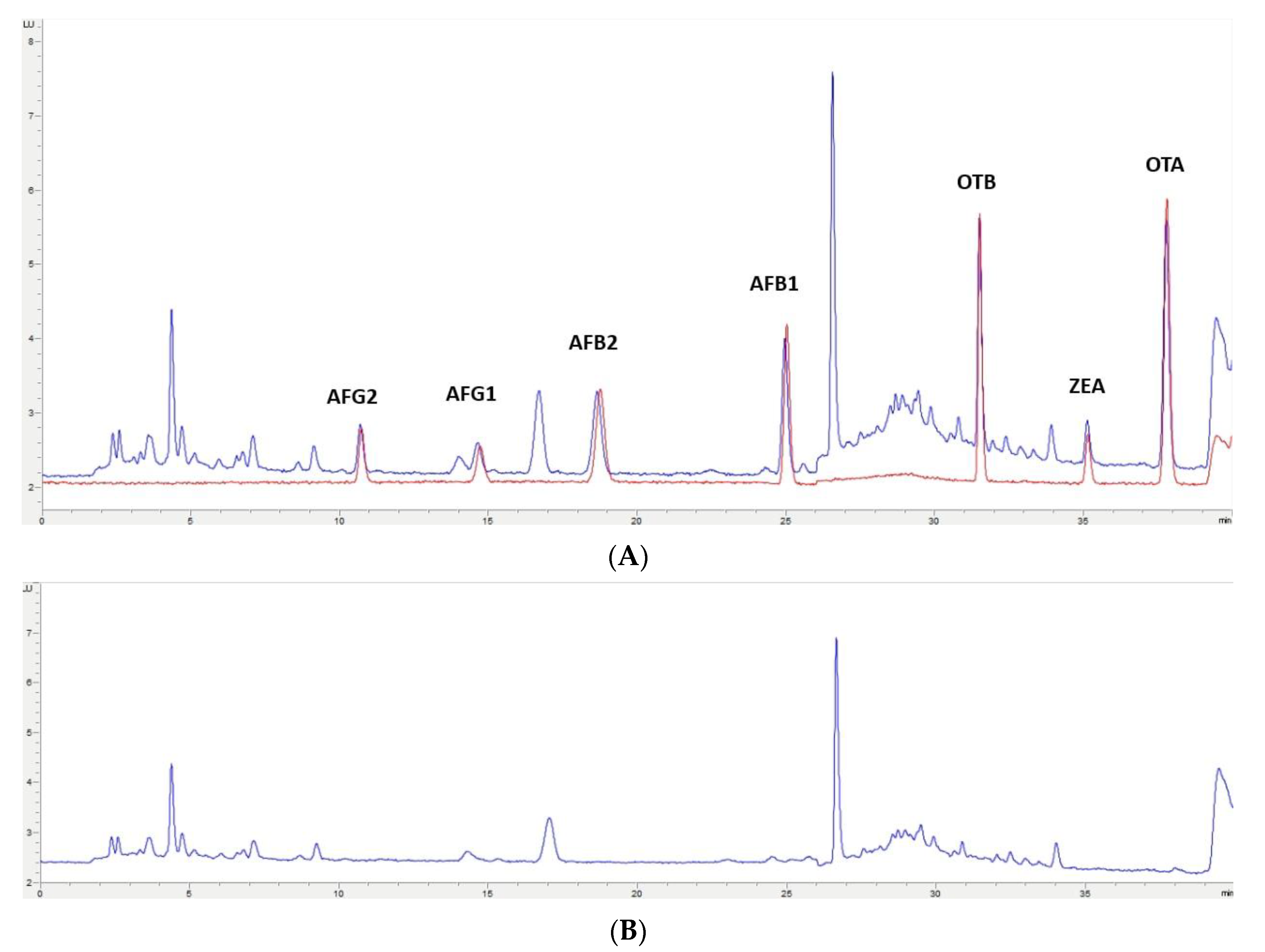
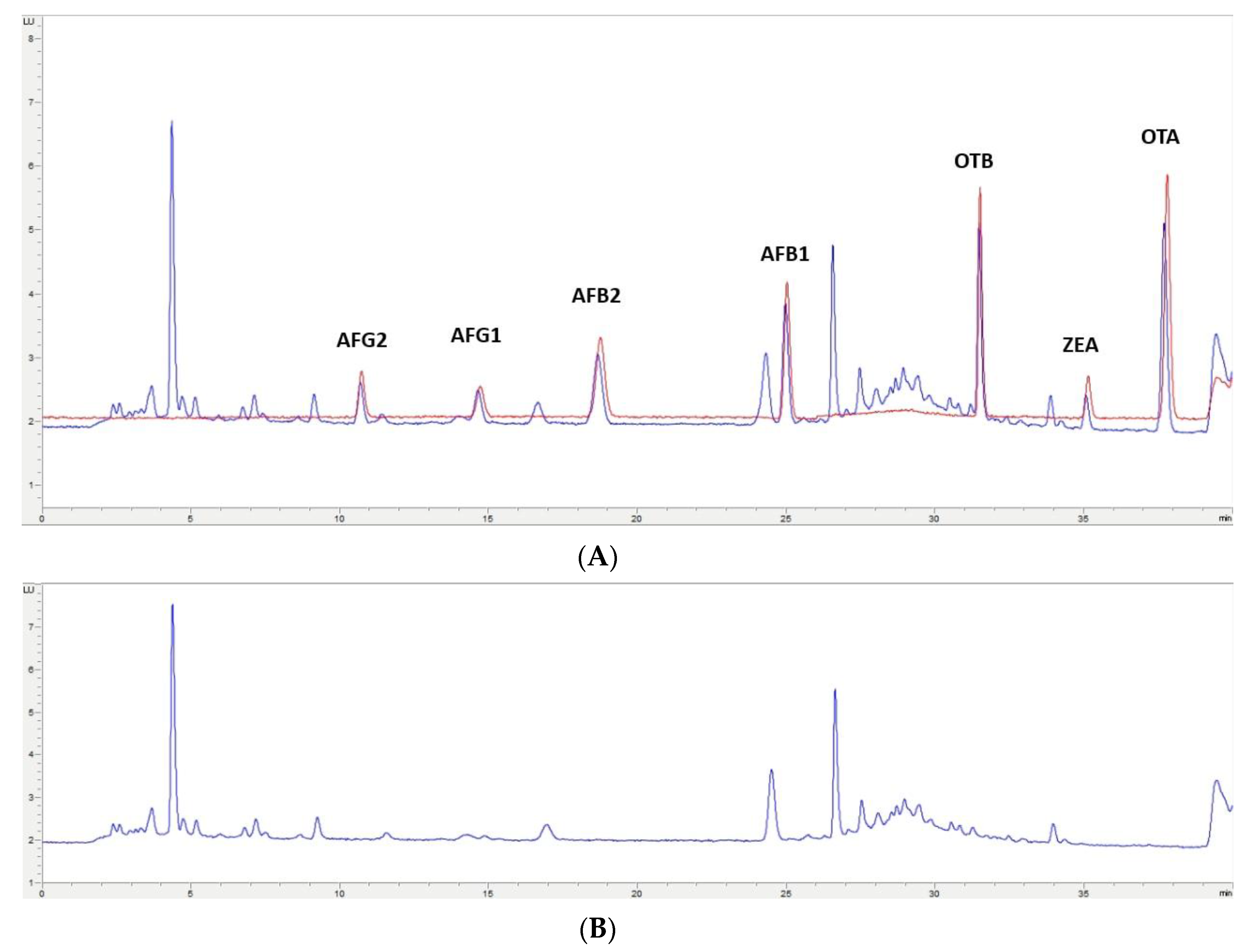
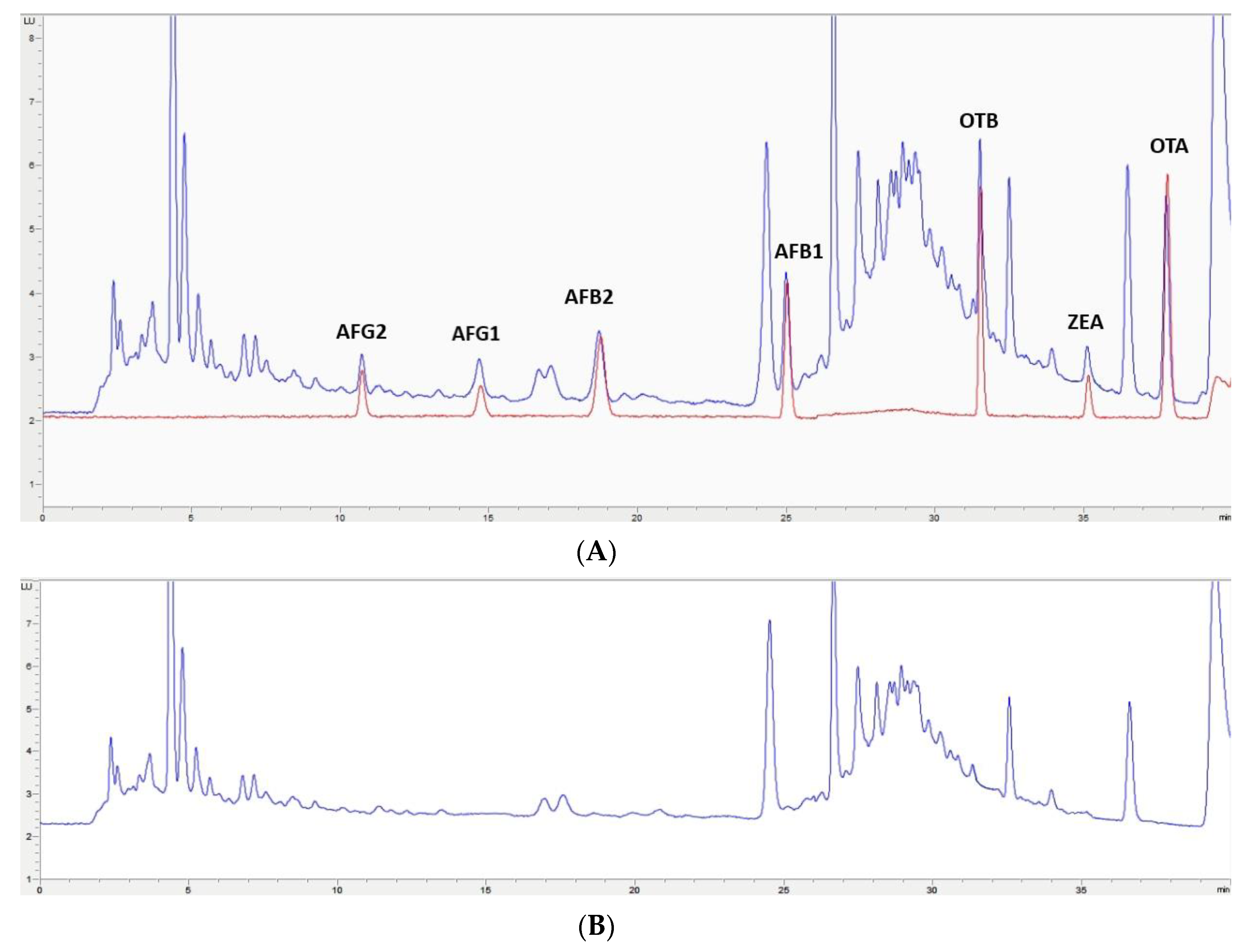
2.4. Application
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Chemical and Reagents
5.2. Mycotoxin Standards
5.3. Safety Precautions
5.4. Animal Feed Samples
5.5. Standard Solutions
5.6. Sample Preparation
5.7. Equipment and Chromatographic Conditions
5.8. Validation of the Analytical Method
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACN | acetonitrile |
| AFB1 | aflatoxin B1 |
| AFB2 | aflatoxin B2 |
| AFG1 | aflatoxin G1 |
| AFG2 | aflatoxin G2 |
| AFs | aflatoxins |
| DLLME | dispersive liquid–liquid microextraction |
| EU | European Union |
| FB1 | fumonisin B1 |
| FB2 | fumonisin B2 |
| FLD | fluorescence detector |
| IAC | immunoaffinity column |
| LC | liquid chromatography |
| LC-MS/MS | LC coupled with MS in tandem |
| LOD | limit of detection |
| LOQ | limit of quantification |
| MeOH | methanol |
| MS | mass spectrometry detector |
| OTA | ochratoxin A |
| OTB | ochratoxin B |
| PBS | phosphate buffer |
| QuEChERS | Quick, Easy, Cheap, Effective, Rugged and Safe |
| rpm | revolutions per minute |
| RE | relative error |
| RSD | relative standard deviation |
| SPE | solid-phase extraction |
| TFA | trifluoroacetic acid |
| ZEA | zearalenone |
References
- European Commission Animal Feed. Available online: https://ec.europa.eu/food/safety/animal-feed_en (accessed on 2 March 2020).
- European Parliament and Council. European Commission Regulation (EC) No 178/2002 of 28 January 2002 laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety. Off. J. Eur. Communities 2002, L31, 1–24. [Google Scholar]
- Adams, C.A. Nutrition-based health in animal production. Nutr. Res. Rev. 2006, 19, 79–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Stefano, V.; Pitonzo, R.; Cicero, N.; D’Oca, M.C. Mycotoxin contamination of animal feedingstuff: Detoxification by gamma-irradiation and reduction of aflatoxins and ochratoxin A concentrations. Food Addit. Contam. Part. A 2014, 31, 2034–2039. [Google Scholar] [CrossRef]
- Arroyo-Manzanares, N.; Huertas-Pérez, J.F.; García-Campaña, A.M.; Gámiz-Gracia, L. Aflatoxins in animal feeds: A straightforward and cost-effective analytical method. Food Control 2015, 54, 74–78. [Google Scholar] [CrossRef]
- Streit, E.; Naehrer, K.; Rodrigues, I.; Schatzmayr, G. Mycotoxin occurrence in feed and feed raw materials worldwide: Long-term analysis with special focus on Europe and Asia. J. Sci. Food Agric. 2013, 93, 2892–2899. [Google Scholar] [CrossRef]
- Sforza, S.; Dall’Asta, C.; Marchelli, R. Recent advances in mycotoxin determination in food and feed by hyphenated chromatographic techniques/mass spectrometry. Mass Spectrom. Rev. 2006, 25, 54–76. [Google Scholar] [CrossRef] [PubMed]
- Eskola, M.; Kos, G.; Elliott, C.T.; Hajšlová, J.; Mayar, S.; Krska, R. Worldwide contamination of food-crops with mycotoxins: Validity of the widely cited ’FAO estimate’ of 25%. Crit. Rev. Food Sci. Nutr. 2019, 3, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kosicki, R.; Błajet-Kosicka, A.; Grajewski, J.; Twarużek, M. Multiannual mycotoxin survey in feed materials and feedingstuffs. Anim. Feed Sci. Technol. 2016, 215, 165–180. [Google Scholar] [CrossRef]
- da Rocha, M.E.B.; Freire, F.D.C.O.; Maia, F.E.F.; Guedes, M.I.F.; Rondina, D. Mycotoxins and their effects on human and animal health. Food Control 2014, 36, 159–165. [Google Scholar] [CrossRef]
- Santos Pereira, C.; Cunha, S.C.; Fernandes, J.O. Prevalent Mycotoxins in Animal Feed: Occurrence and Analytical Methods. Toxins (Basel) 2019, 11, 290. [Google Scholar] [CrossRef] [Green Version]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Chemical agents and related occupations. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 9–562. [Google Scholar]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; Kujawa, M. Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Aromatic Amines and Mycotoxins. IARC Monogr. Eval. Carcinog. Risks Hum. 1994, 56, 351. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer (IARC). Overall Evaluations of Carcinogenicity: An Updating of IARC Monographs Volumes 1 to 42; IARC Monographs on the Identification of Carcinogenic Hazards to Humans Supplement 7; International Agency for Research on Cancer (IARC): Lyon, France, 1987; p. 46. [Google Scholar]
- Jia, R.; Ma, Q.; Fan, Y.; Ji, C.; Zhang, J.; Liu, T.; Zhao, L. The toxic effects of combined aflatoxins and zearalenone in naturally contaminated diets on laying performance, egg quality and mycotoxins residues in eggs of layers and the protective effect of Bacillus subtilis biodegradation product. Food Chem. Toxicol. 2016, 90, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Arroyo-Manzanares, N.; Rodríguez-Estévez, V.; Arenas-Fernández, P.; García-Campaña, A.M.; Gámiz-Gracia, L. Occurrence of Mycotoxins in Swine Feeding from Spain. Toxins (Basel) 2019, 11, 342. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, I.; Handl, J.; Binder, E.M. Mycotoxin occurrence in commodities, feeds and feed ingredients sourced in the Middle East and Africa. Food Addit. Contam. Part B 2011, 4, 168–179. [Google Scholar] [CrossRef] [Green Version]
- European Commission. 2002/32/EC. Directive 2002/32/EC of The European Parliament and of The Council of 7 May 2002 on undesirable substances in animal feed. Off. J. Eur. Union 2002, 140, 12. [Google Scholar]
- European Commission. Commission Recommendation of 17 August 2006 on the presence of deoxynivalenol, zearalenone, ochratoxin A, T-2 and HT-2 and fumonisins in products intended for animal feeding. Off. J. Eur. Union 2006, 229, 7. [Google Scholar]
- Anklam, E.; Stroka, J.; Boenke, A. Acceptance of analytical methods for implementation of EU legislation with a focus on mycotoxins. Food Control 2002, 13, 173–183. [Google Scholar] [CrossRef]
- Selvaraj, J.N.; Zhou, L.; Wang, Y.; Zhao, Y.; Xing, F.; Dai, X.; Liu, Y. Mycotoxin detection—Recent trends at global level. J. Integr. Agric. 2015, 14, 2265–2281. [Google Scholar] [CrossRef]
- Romera, D.; Mateo, E.M.; Mateo-Castro, R.; Gómez, J.V.; Gimeno-Adelantado, J.V.; Jiménez, M. Determination of multiple mycotoxins in feedstuffs by combined use of UPLC–MS/MS and UPLC–QTOF–MS. Food Chem. 2018, 267, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Grajewski, J.; Błajet-Kosicka, A.; Twarużek, M.; Kosicki, R. Occurrence of mycotoxins in Polish animal feed in years 2006–2009. J. Anim. Physiol. Anim. Nutr. (Berlin) 2012, 96, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, M.J.; Kim, H.J.; Cho, S.K.; Park, H.J.; Jeong, M.H. Analytical method development and monitoring of Aflatoxin B1, B2, G1, G2 and Ochratoxin A in animal feed using HPLC with Fluorescence detector and photochemical reaction device. Cogent Food Agric. 2017, 3, 1–8. [Google Scholar] [CrossRef]
- Rahmani, A.; Jinap, S.; Soleimany, F. Validation of the procedure for the simultaneous determination of aflatoxins ochratoxin A and zearalenone in cereals using HPLC-FLD. Food Addit. Contam. Part A 2010, 27, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- Soleimany, F.; Jinap, S.; Rahmani, A.; Khatib, A. Simultaneous detection of 12 mycotoxins in cereals using RP-HPLC-PDA-FLD with PHRED and a post-column derivatization system. Food Addit. Contam. Part A 2011, 28, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Krska, R.; Welzig, E.; Berthiller, F.; Molinelli, A.; Mizaikoff, B. Advances in the analysis of mycotoxins and its quality assurance. Food Addit. Contam. 2005, 22, 345–353. [Google Scholar] [CrossRef]
- Arce-López, B.; Lizarraga, E.; Vettorazzi, A.; González-Peñas, E. Human biomonitoring of mycotoxins in blood, plasma and serum in recent years: a review. Toxins. 2020, 12, 147. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Xu, J.-J.; Huang, B.-F.; Cai, Z.-X.; Ren, Y.-P. High-performance liquid chromatographic determination of multi-mycotoxin in cereals and bean foodstuffs using interference-removal solid-phase extraction combined with optimized dispersive liquid-liquid microextraction. J. Sep. Sci. 2017, 40, 2141–2150. [Google Scholar] [CrossRef]
- Zhao, J.; Zhu, Y.; Jiao, Y.; Ning, J.; Yang, Y. Ionic-liquid-based dispersive liquid–liquid microextraction combined with magnetic solid-phase extraction for the determination of aflatoxins B1, B2, G1, and G2 in animal feeds by high-performance liquid chromatography with fluorescence detection. J. Sep. Sci. 2016, 39, 3789–3797. [Google Scholar] [CrossRef]
- Kralj Cigić, I.; Prosen, H. An Overview of Conventional and Emerging Analytical Methods for the Determination of Mycotoxins. Int. J. Mol. Sci. 2009, 10, 62–115. [Google Scholar] [CrossRef]
- Smith, L.L.; Francis, K.A.; Johnson, J.T.; Gaskill, C.L. Quantitation of fumonisin B1 and B2 in feed using FMOC pre-column derivatization with HPLC and fluorescence detection. Food Chem. 2017, 234, 174–179. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, S.; Zheng, H.; He, C.; Zhang, H. T-2 toxin, zearalenone and fumonisin B 1 in feedstuffs from China. Food Addit. Contam. Part B 2013, 6, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Khayoon, W.S.; Saad, B.; Salleh, B.; Ismail, N.A.; Manaf, N.H.A.; Latiff, A.A. A reversed phase high performance liquid chromatography method for the determination of fumonisins B1 and B2 in food and feed using monolithic column and positive confirmation by liquid chromatography/tandem mass spectrometry. Anal. Chim. Acta 2010, 679, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Kaltner, F.; Rampl, C.; Rychlik, M.; Zimmermann, T.; Rohe, A. Development and Validation of a Cost-Effective HPLC-FLD Method for Routine Analysis of Fumonisins B1 and B2 in Corn and Corn Products. Food Anal. Methods 2017, 10, 1349–1358. [Google Scholar] [CrossRef]
- Feizy, J.; Beheshti, H.R.; Asadi, M. A survey of aflatoxin in cotton seed in Iran by HPLC with on-line photochemical derivatisation and fluorescence detection. Food Addit. Contam. Part B Surveill. 2012, 5, 200–203. [Google Scholar] [CrossRef]
- Asghar, M.A.; Iqbal, J.; Ahmed, A.; Khan, M.A.; Shamsuddin, Z.A.; Jamil, K. Development and validation of a high-performance liquid chromatography method with post-column derivatization for the detection of aflatoxins in cereals and grains. Toxicol. Ind. Health 2016, 32, 1122–1134. [Google Scholar] [CrossRef]
- Ibáñez-Vea, M.; Corcuera, L.A.; Remiro, R.; Murillo-Arbizu, M.T.; González-Peñas, E.; Lizarraga, E. Validation of a UHPLC-FLD method for the simultaneous quantification of aflatoxins, ochratoxin A and zearalenone in barley. Food Chem. 2011, 127, 351–358. [Google Scholar] [CrossRef]
- Lee, M.; Seo, D.J.; Jeon, S.B.; Ok, H.E.; Jung, H.; Choi, C.; Chun, H.S. Detection of Foodborne Pathogens and Mycotoxins in Eggs and Chicken Feeds from Farms to Retail Markets. Korean J. Food Sci. Anim. Resour. 2016, 36, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Khayoon, W.S.; Saad, B.; Yan, C.B.; Hashim, N.H.; Ali, A.S.M.; Salleh, M.I.; Salleh, B. Determination of aflatoxins in animal feeds by HPLC with multifunctional column clean-up. Food Chem. 2010, 118, 882–886. [Google Scholar] [CrossRef]
- Njobeh, P.B.; Dutton, M.F.; Åberg, A.T.; Haggblom, P. Estimation of Multi-Mycotoxin Contamination in South African Compound Feeds. Toxins (Basel). 2012, 4, 836–848. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, S.Z.; Asi, M.R.; Nisar, S.; Zia, K.M.; Jinap, S.; Malik, N. A Limited Survey of Aflatoxins and Zearalenone in Feed and Feed Ingredients from Pakistan. J. Food Prot. 2016, 79, 1798–1801. [Google Scholar] [CrossRef]
- Sahin, H.Z.; Celik, M.; Kotay, S.; Kabak, B. Aflatoxins in dairy cow feed, raw milk and milk products from Turkey. Food Addit. Contam. Part B 2016, 9, 152–158. [Google Scholar] [CrossRef]
- Li, X.; Zhao, L.; Fan, Y.; Jia, Y.; Sun, L.; Ma, S.; Ji, C.; Ma, Q.; Zhang, J. Occurrence of mycotoxins in feed ingredients and complete feeds obtained from the Beijing region of China. J. Anim. Sci. Biotechnol. 2014, 5, 37. [Google Scholar] [CrossRef] [Green Version]
- Martins, H.M.; Almeida, I.; Camacho, C.; Costa, J.M.; Bernardo, F. A survey on the occurrence of ochratoxin A in feeds for swine and laying hens. Mycotoxin Res. 2012, 28, 107–110. [Google Scholar] [CrossRef]
- Pietruszka, K.; Piątkowska, M.; Jedziniak, P. Occurrence of ochratoxin A in animal tissues and feeds in Poland in 2014–2016. J. Vet. Res. 2017, 61, 483–487. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Li, J.; Li, Y.; Li, T.; He, Q.; Tang, Y.; Liu, H.; Su, Y.; Yin, Y.; Liao, P. Aflatoxin B1, zearalenone and deoxynivalenol in feed ingredients and complete feed from different Province in China. J. Anim. Sci. Biotechnol. 2016, 7, 63. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Sun, L.; Zhang, J.; Guo, J.; Chen, L.; Qi, D.; Zhang, N. Aflatoxin B1, zearalenone and deoxynivalenol in feed ingredients and complete feed from central China. Food Addit. Contam. Part B 2016, 9, 91–97. [Google Scholar] [CrossRef]
- Dimitrieska-Stojković, E.; Stojanovska-Dimzoska, B.; Ilievska, G.; Uzunov, R.; Stojković, G.; Hajrulai-Musliu, Z.; Jankuloski, D. Assessment of aflatoxin contamination in raw milk and feed in Macedonia during 2013. Food Control. 2016, 59, 201–206. [Google Scholar] [CrossRef]
- European Commission. Commission Recommendation 401/2006 of 23 February 2006 laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs. Off. J. Eur. Union 2006, 70, 12–34. [Google Scholar]
- European Commission. Guidance Document on Identification of Mycotoxins in Food and Feed. SANTE/12089/2016. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/cs_contaminants_sampling_guid-doc-ident-mycotoxins.pdf (accessed on 2 March 2020).
- European Commission. Commission decision of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results (2002/657/EC). Off. J. Eur. Communities 2002, 221, 8–36. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mycotoxin | Range (ng/mL) | Curve Equation | R2 | (Slope CI p = 95%) | LOD (µg/kg) | RE (%) of B.C. |
|---|---|---|---|---|---|---|
| AFB1 | 0.4–4 | y = 23.71x + 0.91 | 0.9993 | 22.99–24.43 | 2 | <5 |
| AFB2 | 0.126–1.26 | y = 58.69x + 0.42 | 0.9992 | 56.81–60.56 | 0.63 | <6 |
| AFG1 | 0.4–4 | y = 7.80x + 0.09 | 0.9993 | 7.56–9.04 | 2 | <5 |
| AFG2 | 0.126–1.26 | y = 18.88x + 0.37 | 0.9920 | 18.28–19.49 | 0.63 | <6 |
| OTA | 1–10 | y = 7.32x + 1.20 | 0.9994 | 7.11–7.54 | 5 | <4 |
| OTB | 1–10 | y = 6.23x + 0.98 | 0.9976 | 5.88–6.58 | 5 | <7 |
| ZEA | 8.4–84 | y = 0.25x + 1.65 | 0.9971 | 0.24–0.27 | 42 | <14 |
| Mycotoxin | Precision (%RSD) | Trueness (%RE) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Within-Day (n = 3) | Between-Day (n = 9) | Within-Day (n = 3) | Between-Day (n = 9) | |||||||||
| LOQ | 2.5× LOQ | 10× LOQ | LOQ | 2.5× LOQ | 10× LOQ | LOQ | 2.5× LOQ | 10× OQ | LOQ | 2.5× LOQ | 10× LOQ | |
| AFB1 | 2.1 | 2.8 | 2.8 | 2.6 | 2.1 | 1.8 | 3.6 | 1.5 | 0.7 | 2.1 | 2.7 | 0.7 |
| AFB2 | 3.1 | 2.1 | 3.1 | 2.6 | 1.9 | 2,0 | 4.2 | 0.2 | 0.9 | 1.6 | 1.0 | 0.7 |
| AFG1 | 2.3 | 2.9 | 2.4 | 4.1 | 3.8 | 1.5 | 4.3 | 0.4 | 0.6 | 3.1 | 0.8 | 0.5 |
| AFG2 | 0.9 | 0.3 | 0.2 | 1.7 | 1.8 | 0.6 | 1.7 | 0.3 | 0.6 | 2.4 | 0.1 | 0.7 |
| OTA | 0.4 | 2.3 | 3.7 | 4.5 | 2.3 | 2.4 | 3.6 | 1.7 | 0.8 | 4.6 | 1.7 | 1.1 |
| OTB | 2.6 | 0.3 | 0.4 | 2.2 | 1.9 | 0.8 | 5.7 | 1.8 | 1.4 | 6.2 | 1.5 | 1.3 |
| ZEA | 4.3 | 3.8 | 4.9 | 7.3 | 4.2 | 3.0 | 13.4 | 0.7 | 1.5 | 5.7 | 0.5 | 0.8 |
| Mycotoxin | Recovery (n = 3; %RSD) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Poultry | Pigs | Cows | Sheep | |||||||||
| LOQ | 2.5× LOQ | 5× LOQ | LOQ | 2.5× LOQ | 5× LOQ | LOQ | 2.5× LOQ | 5× LOQ | LOQ | 2.5× LOQ | 5× LOQ | |
| AFB1 | 78.5 (4.4) | 80.8 (0.9) | 77.9 (1.7) | 82.3 (2.0) | 84.2 (1.7) | 86.6 (0.8) | 82.5 (1.5) | 81.3 (0.6) | 82.3 (1.1) | 83.5 (0.9) | 86.4 (1.3) | 80.7 (0.3) |
| AFB2 | 87,0 (2.8) | 88.4 (0.4) | 79.2 (0.1) | 88.3 (2.1) | 89.4 (1.0) | 84.7 (1.1) | 85.5 (0.5) | 89.6 (1.0) | 78.9 (0.7) | 88.3 (0.3) | 89.1 (0.6) | 79.7 (1.4) |
| AFG1 | 73.0 (5.1) | 72.7 (3.5) | 75.5 (2.0) | 80.7 (1.2) | 75.8 (0.2) | 76.2 (0.7) | 70.0 (1.3) | 75.5 (1.3) | 72.4 (0.5) | 76.7(0.9) | 79.3 (1.5) | 74.6 (0.3) |
| AFG2 | 86.1 (3.3) | 81.6 (2,0) | 81.6 (1.0) | 74.2 (2.6) | 79.3 (3.2) | 84.7 (0.3) | 82.6 (1.1) | 84.6 (1.5) | 78.9 (0.2) | 89.5 (0.5) | 81.3 (1.2) | 78.6 (0.3) |
| OTA | 83.4 (3.1) | 80.4 (1.1) | 79.5 (1.0) | 87.5 (2.3) | 84.6 (1.6) | 87.1 (1.5) | 83.8 (1.9) | 86.5 (2.0) | 81.7 (1.7) | 85.8 (2.6) | 82.4 (5.0) | 81.1 (0.4) |
| OTB | 82.4 (3.9) | 85.1 (0.2) | 74.9 (0.8) | 85.1 (3.4) | 75.6 (0.9) | 77.6 (1.3) | 79.5 (3.3) | 76.4 (4.7) | 75.0 (2.1) | 76.6 (2.1) | 74.2 (1.6) | 73.4 (0.7) |
| ZEA | 79.0 (0.8) | 74.3 (3.0) | 76.6 (0.6) | 73.3 (1.3) | 72.9 (4.2) | 77.0 (1.0) | 71.2 (3.8) | 73.7 (1.6) | 77.8 (0.7) | 75.2 (4.6) | 73.0 (1.6) | 80.2 (0.5) |
| Mycotoxin | Recovery Between-Day (n = 9; %RSD) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Poultry | Pigs | Cows | Sheep | |||||||||
| LOQ | 2.5× LOQ | 5× LOQ | LOQ | 2.5× LOQ | 5× LOQ | LOQ | 2.5× LOQ | 5× LOQ | LOQ | 2.5× LOQ | 5× LOQ | |
| AFB1 | 81.1 (3.5) | 81.7 (1.4) | 77.1 (1.6) | 82.9 (1.3) | 81.9 (2.4) | 84.8 (1.8) | 82.6 (1.2) | 83.8 (2.4) | 83.1 (1.2) | 82.3 (2.0) | 85.6 (1.2) | 81.4 (1.2) |
| AFB2 | 87.0 (2.5) | 88.4 (1.0) | 79.2 (2.1) | 88.8 (1.9) | 91.8 (2.4) | 83.4 (1.7) | 86.4 (1.6) | 92.7 (2.6) | 79.2 (1.0) | 86.9 (1.8) | 90.7 (3.3) | 80.1 (0.9) |
| AFG1 | 73.0 (3.1) | 72.7 (4.2) | 75.5 (2.3) | 77.3 (6.2) | 74.9 (2.3) | 75.7 (1.1) | 72.6 (3.8) | 74.6 (2.8) | 73.6 (2.8) | 75.7 (2.0) | 78.0 (2.0) | 75.6 (2.8) |
| AFG2 | 86.1 (4.4) | 81.6 (2.2) | 81.6 (1.7) | 78.5 (4.6) | 79.1 (2.8) | 83.3 (1.8) | 83.6 (4.9) | 83.7 (1.9) | 78.5 (1.4) | 86.4 (5.2) | 80.7 (2.2) | 78.9 (1.2) |
| OTA | 83.4 (2.3) | 80.4 (2.0) | 79.5 (2.1) | 86.0 (2.4) | 85.5 (1.8) | 85.8 (2.8) | 82.9 (2.2) | 86.6 (1.5) | 81.2 (1.0) | 85.7 (2.0) | 82.9 (2.7) | 80.5 (1.7) |
| OTB | 82.4 (4.4) | 85.1 (3.0) | 74.9 (1.8) | 84.2 (3.2) | 76.8 (1.6) | 78.4 (1.4) | 79.6 (3.2) | 77.3 (2.6) | 76.2 (1.6) | 77.0 (2.7) | 75.5 (1.8) | 76.1 (2.9) |
| ZEA | 79.0 (3.6) | 74.3 (1.7) | 76.6 (0.8) | 75.6 (3.0) | 73.5 (2.3) | 77.8 (1.8) | 73.6 (3.8) | 73.7 (1.8) | 77.8 (0.8) | 75.2 (3.0) | 72.9 (2.1) | 78.9 (1.7) |
| Mycotoxin | Poultry (n = 27; RSD%) | Pigs (n = 27; RSD%) | Cows (n = 27; RSD%) | Sheep (n = 27; RSD%) |
|---|---|---|---|---|
| AFB1 | 80.0 (3.4) | 83.2 (2.3) | 83.2 (1.7) | 83.1 (2.7) |
| AFB2 | 84.9 (5.2) | 88.0 (4.4) | 86.1 (6.8) | 85.9 (5.7) |
| AFG1 | 73.6 (3.6) | 75.9 (4.0) | 73.6 (3.2) | 76.4 (2.7) |
| AFG2 | 83.1 (3.9) | 80.3 (4.1) | 81.9 (4.3) | 82.0 (5.2) |
| OTA | 82.6 (3.8) | 85.8 (2.3) | 83.6 (3.2) | 83.0 (3.4) |
| OTB | 79.2 (5.0) | 79.8 (4.6) | 77.7 (3.1) | 76.2 (2.5) |
| ZEA | 76.7 (3.4) | 75.6 (3.3) | 75.1 (3.5) | 75.7 (4.0) |
| Type of Feed | Protein% | Fat% | Ash% | Crude Fiber% |
|---|---|---|---|---|
| Cows | 11.14 | 4.14 | 5.04 | 7.22 |
| Sheep | 10.89 | 3.01 | 6.22 | 12.20 |
| Pigs | 20.21 | 4.61 | 9.66 | 8.82 |
| Poultry | 15.34 | 5.09 | 4.65 | 3.89 |
| Time (min) | %Acidified Water | %ACN | %MeOH |
|---|---|---|---|
| 0.00 | 70.0 | 9.0 | 21.0 |
| 12.55 | 73.0 | 9.0 | 18.0 |
| 21.70 | 73.0 | 9.0 | 18.0 |
| 21.75 | 65.0 | 9.0 | 26.0 |
| 22.50 | 65.0 | 9.0 | 26.0 |
| 26.50 | 45.0 | 16.5 | 38.5 |
| 36.80 | 45.0 | 16.5 | 38.5 |
| 36.85 | 10.0 | 45.0 | 45.0 |
| 44.00 | 10.0 | 45.0 | 45.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz-Solano, B.; González-Peñas, E. Mycotoxin Determination in Animal Feed: An LC-FLD Method for Simultaneous Quantification of Aflatoxins, Ochratoxins and Zearelanone in This Matrix. Toxins 2020, 12, 374. https://doi.org/10.3390/toxins12060374
Muñoz-Solano B, González-Peñas E. Mycotoxin Determination in Animal Feed: An LC-FLD Method for Simultaneous Quantification of Aflatoxins, Ochratoxins and Zearelanone in This Matrix. Toxins. 2020; 12(6):374. https://doi.org/10.3390/toxins12060374
Chicago/Turabian StyleMuñoz-Solano, Borja, and Elena González-Peñas. 2020. "Mycotoxin Determination in Animal Feed: An LC-FLD Method for Simultaneous Quantification of Aflatoxins, Ochratoxins and Zearelanone in This Matrix" Toxins 12, no. 6: 374. https://doi.org/10.3390/toxins12060374
APA StyleMuñoz-Solano, B., & González-Peñas, E. (2020). Mycotoxin Determination in Animal Feed: An LC-FLD Method for Simultaneous Quantification of Aflatoxins, Ochratoxins and Zearelanone in This Matrix. Toxins, 12(6), 374. https://doi.org/10.3390/toxins12060374