Crystal Structure of Exotoxin A from Aeromonas Pathogenic Species


Abstract
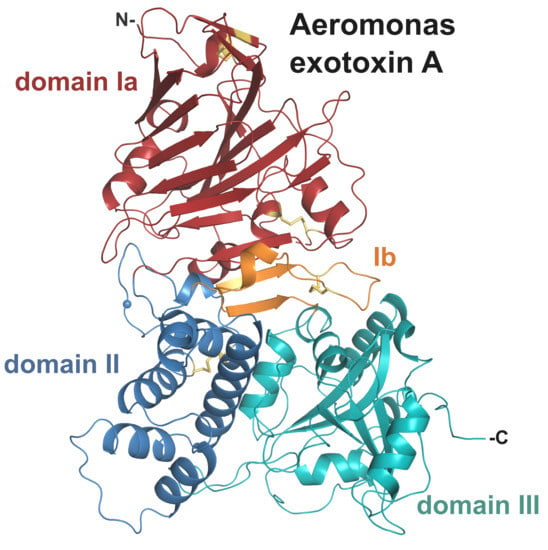
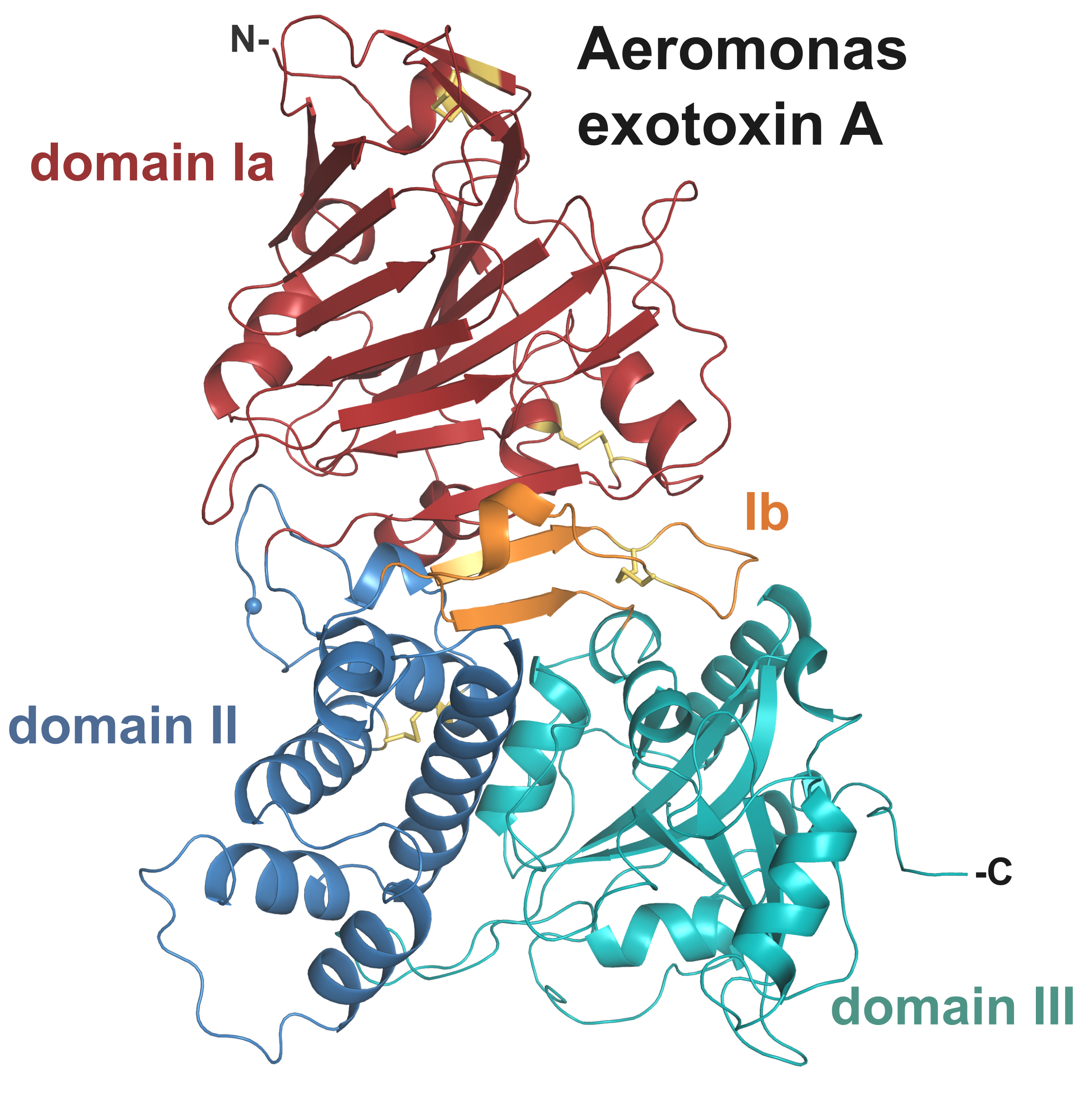
:
1. Introduction
2. Results and Discussion
2.1. Genomic Data Mining Suggests AE is Common to Several Aeromonas Species
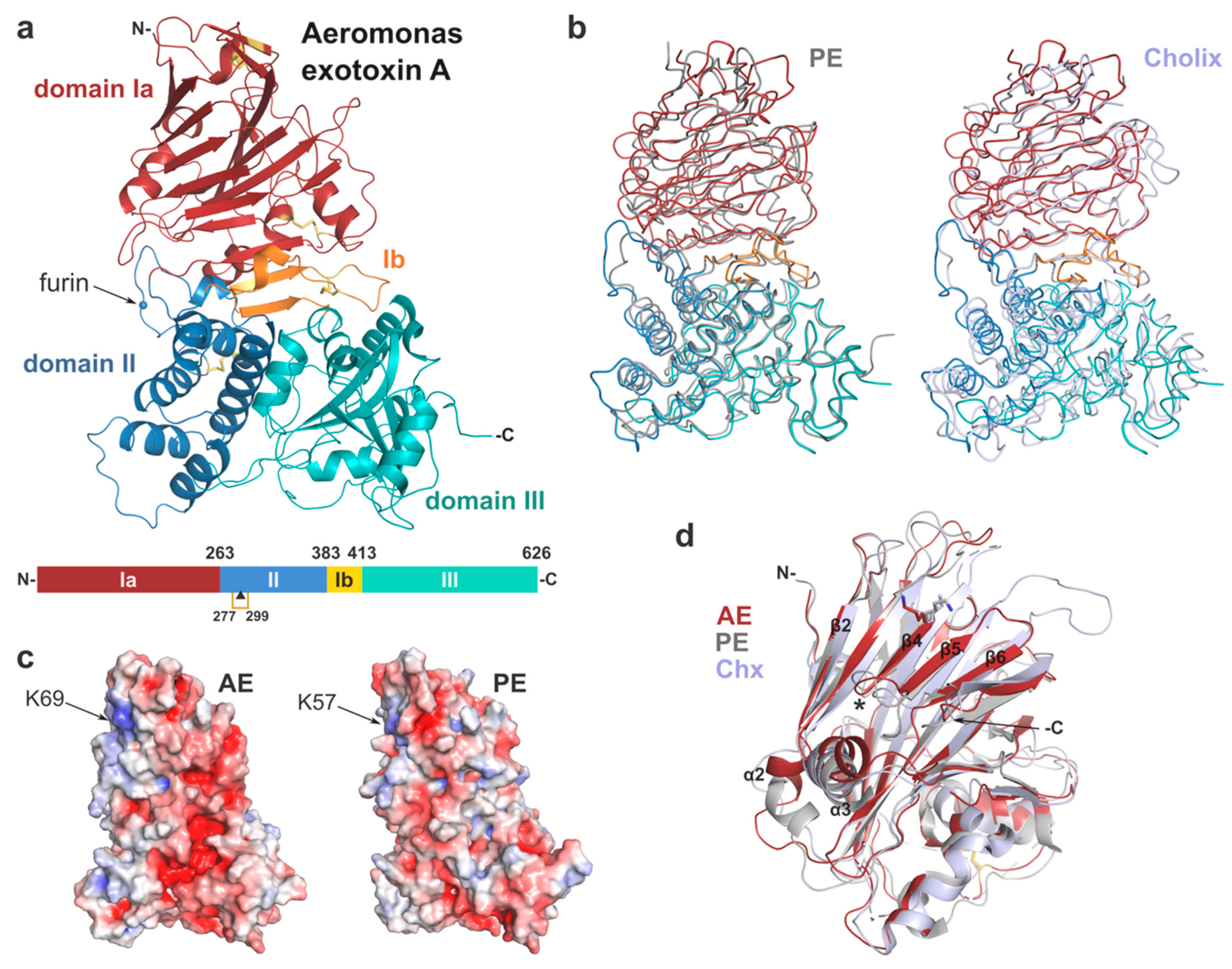
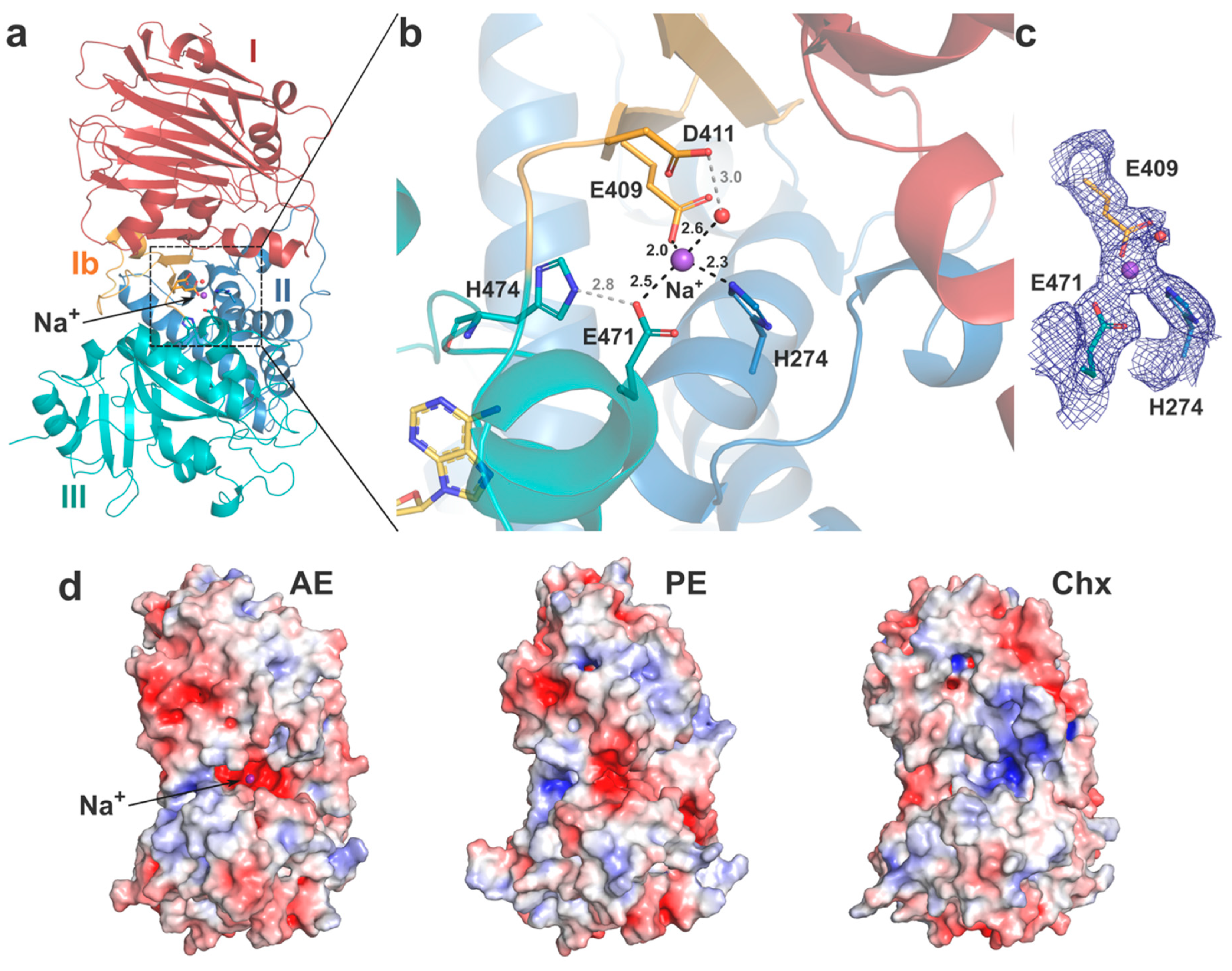
2.2. Crystal Structure of AE
2.2.1. Domain I and Implications for Receptor Binding
2.2.2. Structural Elements Involved in Intracellular Trafficking
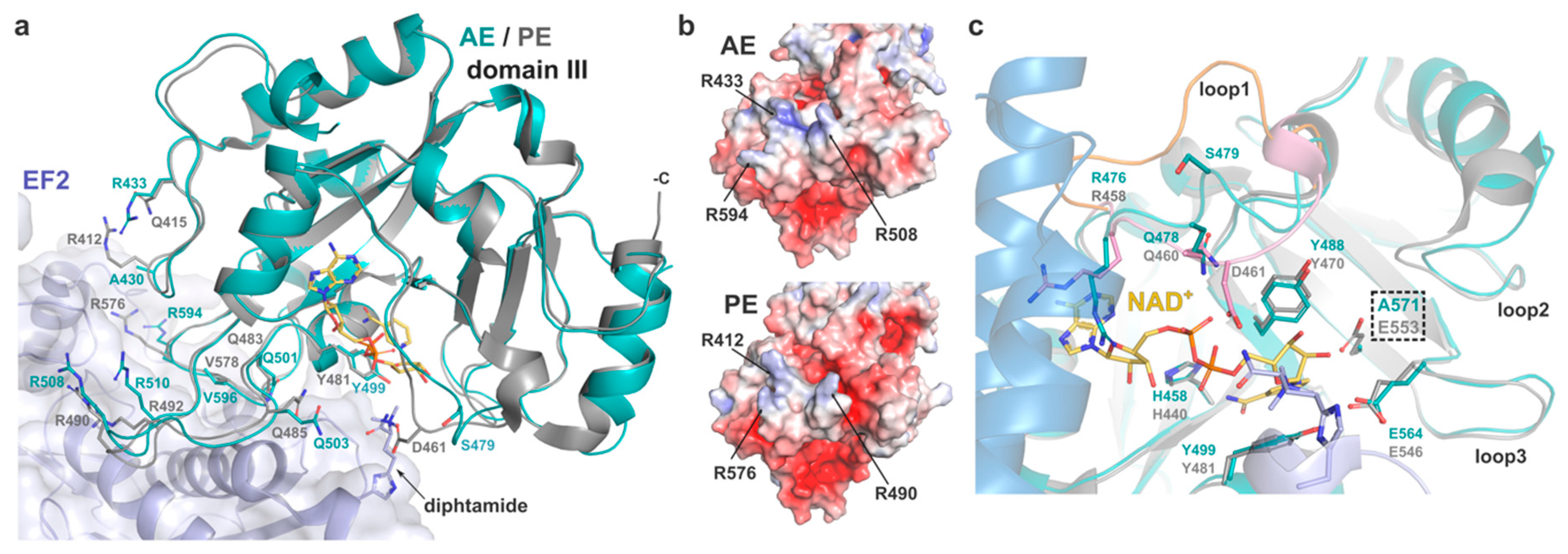
2.2.3. Domain III and Implications for ADP-Ribosylating Activity on eEF2
2.2.4. Catalytic Site
2.2.5. Interdomain Interactions
3. Conclusions
4. Materials and Methods
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Lugo, M.R.; Merrill, A.R. The Father, Son and Cholix Toxin: The Third Member of the DT Group Mono-ADP-Ribosyltransferase Toxin Family. Toxins 2015, 7, 2757–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, B.A.; Collier, R.J. Diphtheria Toxin and Pseudomonas aeruginosa Exotoxin A: Active-Site Structure and Enzymic Mechanism. Curr. Top. Microbiol. Immunol. 1992, 175, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, R.; Wang, Y.; Visschedyk, D.; Merrill, A.R. The nature and character of the transition state for the ADP-ribosyltransferase reaction. EMBO Rep. 2008, 9, 802–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kounnas, M.Z.; E Morris, R.; Thompson, M.R.; Fitzgerald, D.J.; Strickland, D.K.; Saelinger, C.B. The alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein binds and internalizes Pseudomonas exotoxin A. J. Biol. Chem. 1992, 267, 12420–12423. [Google Scholar] [PubMed]
- Jørgensen, R.; Purdy, A.E.; Fieldhouse, R.J.; Kimber, M.S.; Bartlett, D.H.; Merrill, A.R. Cholix Toxin, a Novel ADP-ribosylating Factor from Vibrio cholerae. J. Biol. Chem. 2008, 283, 10671–10678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, M.; Fryling, C.M.; Pastan, I.; Fitzgerald, D.J. Cell-mediated cleavage of Pseudomonas exotoxin between Arg279 and Gly280 generates the enzymatically active fragment which translocates to the cytosol. J. Biol. Chem. 1992, 267, 25396–25401. [Google Scholar]
- Jørgensen, R.; Merrill, A.R.; Yates, S.P.; Marquez, V.E.; Schwan, A.; Boesen, T.; Andersen, G.R. Exotoxin A–eEF2 complex structure indicates ADP ribosylation by ribosome mimicry. Nature 2005, 436, 979–984. [Google Scholar] [CrossRef]
- Pastan, I.; Fitzgerald, D. Pseudomonas exotoxin: Chimeric toxins. J. Biol. Chem. 1989, 264, 15157–15160. [Google Scholar]
- Kreitman, R.J.; Dearden, C.; Zinzani, P.L.; Delgado, J.; Karlin, L.; Robak, T.; Gladstone, D.; Le Coutre, P.; Dietrich, S.; Gotic, M.; et al. Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia. Leukemia 2018, 32, 1768–1777. [Google Scholar] [CrossRef]
- Mrsny, R.J.; Daugherty, A.L.; McKee, M.L.; Fitzgerald, D.J. Bacterial toxins as tools for mucosal vaccination. Drug Discov. Today 2002, 7, 247–258. [Google Scholar] [CrossRef]
- Taverner, A.; Mackay, J.; Laurent, F.; Hunter, T.; Liu, K.; Mangat, K.; Song, L.; Seto, E.; Postlethwaite, S.; Alam, A.; et al. Cholix protein domain I functions as a carrier element for efficient apical to basal epithelial transcytosis. Tissue Barriers 2020, 8, 1710429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugherty, A.L.; McKee, M.L.; Fitzgerald, D.J.; Mrsny, R.J. Epithelial application of Pseudomonas aeruginosa exotoxin A results in a selective targeting to cells in the liver, spleen, and lymph node. J. Control. Rel. 2000, 65, 297–302. [Google Scholar] [CrossRef]
- Grim, C.J.; Kozlova, E.V.; Ponnusamy, D.; Fitts, E.C.; Sha, J.; Kirtley, M.L.; van Lier, C.J.; Tiner, B.L.; Erova, T.E.; Joseph, S.J.; et al. Functional Genomic Characterization of Virulence Factors from Necrotizing Fasciitis-Causing Strains of Aeromonas hydrophila. Appl. Environ. Microbiol. 2014, 80, 4162–4183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Bravo, A.; Kilgore, P.B.; Andersson, J.A.; Blears, E.; Figueras, M.J.; Hasan, N.A.; Colwell, R.R.; Sha, J.; Chopra, A.K. T6SS and ExoA of flesh-eating Aeromonas hydrophila in peritonitis and necrotizing fasciitis during mono- and polymicrobial infections. Proc. Natl. Acad. Sci. USA 2019, 116, 24084–24092. [Google Scholar] [CrossRef] [PubMed]
- Janda, J.M.; Abbott, S.L. The Genus Aeromonas: Taxonomy, Pathogenicity, and Infection. Clin. Microbiol. Rev. 2010, 23, 35–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bello-López, J.M.; Cabrero-Martínez, O.A.; Ibáñez-Cervantes, G.; Cortez, C.H.; Pelcastre-Rodríguez, L.I.; Gonzalez-Avila, L.U.; Castro-Escarpulli, G. Horizontal Gene Transfer and Its Association with Antibiotic Resistance in the Genus Aeromonas spp. Microorganisms 2019, 7, 363. [Google Scholar] [CrossRef] [Green Version]
- Khajanchi, B.K.; Fadl, A.A.; Borchardt, M.A.; Berg, R.L.; Horneman, A.J.; Stemper, M.E.; Joseph, S.W.; Moyer, N.P.; Sha, J.; Chopra, A.K. Distribution of Virulence Factors and Molecular Fingerprinting of Aeromonas Species Isolates from Water and Clinical Samples: Suggestive Evidence of Water-to-Human Transmission. Appl. Environ. Microbiol. 2010, 76, 2313–2325. [Google Scholar] [CrossRef] [Green Version]
- Beaz-Hidalgo, R.; Alperi, A.; Buján, N.; Romalde, J.L.; Figueras, M.J. Comparison of phenotypical and genetic identification of Aeromonas strains isolated from diseased fish. Syst. Appl. Microbiol. 2010, 33, 149–153. [Google Scholar] [CrossRef]
- Tomas, J. The Main Aeromonas Pathogenic Factors. ISRN Microbiol. 2012, 2012, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Bravo, A.; Figueras, M.J. An Update on the Genus Aeromonas: Taxonomy, Epidemiology, and Pathogenicity. Microorganisms 2020, 8, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fieldhouse, R.J.; Merrill, A.R. Needle in the haystack: Structure-based toxin discovery. Trends Biochem. Sci. 2008, 33, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Chiron, M.F.; Fryling, C.M.; Fitzgerald, D.J. Cleavage of Pseudomonas exotoxin and Diphtheria toxin by a furin-like enzyme prepared from beef liver. J. Biol. Chem. 1994, 269, 18167–18176. [Google Scholar] [PubMed]
- Chaudhary, V.K.; Jinno, Y.; Fitzgerald, D.; Pastan, I. Pseudomonas exotoxin contains a specific sequence at the carboxyl terminus that is required for cytotoxicity. Proc. Natl. Acad. Sci. USA 1990, 87, 308–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Jinno, Y.; Chaudhary, V.K.; Kondo, T.; Adhya, S.; Fitzgerald, D.J.; Pastan, I. Mutational analysis of domain I of Pseudomonas exotoxin. Mutations in domain I of Pseudomonas exotoxin which reduce cell binding and animal toxicity. J. Biol. Chem. 1988, 263, 13203–13207. [Google Scholar]
- Chaudry, G.J.; Wilson, R.B.; Draper, R.K.; Clowes, R.C. A dipeptide insertion in domain I of exotoxin A that impairs receptor binding. J. Biol. Chem. 1989, 264, 15151–15156. [Google Scholar]
- Holm, L. Benchmarking fold detection by DaliLite v.5. Bioinformatics 2019, 35, 5326–5327. [Google Scholar] [CrossRef]
- Hwang, J.; Fitzgerald, D.J.; Adhya, S.; Pastan, I. Functional domains of pseudomonas exotoxin identified by deletion analysis of the gene expressed in E. coli. Cell 1987, 48, 129–136. [Google Scholar] [CrossRef]
- Siegall, C.B.; Chaudhary, V.K.; Fitzgerald, D.J.; Pastan, I. Functional analysis of domains II, Ib, and III of Pseudomonas exotoxin. J. Biol. Chem. 1989, 264, 14256–14261. [Google Scholar]
- Inocencio, N.M.; Moehring, J.M.; Moehring, T.J. Furin activates Pseudomonas exotoxin A by specific cleavage in vivo and in vitro. J. Biol. Chem. 1994, 269, 31831–31835. [Google Scholar]
- McKee, M.L.; Fitzgerald, D.J. Reduction of furin-nicked Pseudomonas exotoxin A: An unfolding story. Biochemistry 1999, 38, 16507–16513. [Google Scholar] [CrossRef] [PubMed]
- Weldon, J.; Pastan, I. A guide to taming a toxin: Recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011, 278, 4683–4700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hessler, J.L.; Kreitman, R.J. An Early Step in Pseudomonas Exotoxin Action Is Removal of the Terminal Lysine Residue, Which Allows Binding to the KDEL Receptor. Biochemistry 1997, 36, 14577–14582. [Google Scholar] [CrossRef] [PubMed]
- Raykhel, I.; Alanen, H.; Salo, K.; Jurvansuu, J.; Nguyen, V.D.; Latva-Ranta, M.; Ruddock, L.W. A molecular specificity code for the three mammalian KDEL receptors. J. Cell Biol. 2007, 179, 1193–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreitman, R.J.; Pastan, I. Importance of the glutamate residue of KDEL in increasing the cytotoxicity of Pseudomonas exotoxin derivatives and for increased binding to the KDEL receptor. Biochem. J. 1995, 307, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Koopmann, J.-O.; Albring, J.; Hüter, E.; Bulbuc, N.; Spee, P.; Neefjes, J.; Hämmerling, G.J.; Momburg, F. Export of Antigenic Peptides from the Endoplasmic Reticulum Intersects with Retrograde Protein Translocation through the Sec61p Channel. Immunity 2000, 13, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Lord, J.M.; Roberts, L.M.; Lencer, W.I. Entry of Protein Toxins into Mammalian Cells by Crossing the Endoplasmic Reticulum Membrane: Co-opting Basic Mechanisms of Endoplasmic Reticulum-Associated Degradation. Curr. Top. Microbiol. Immunol. 2005, 300, 149–168. [Google Scholar] [CrossRef]
- Kellner, A.; Taylor, M.; Banerjee, T.; Britt, C.B.; Teter, K. A binding motif for Hsp90 in the A chains of ADP-ribosylating toxins that move from the endoplasmic reticulum to the cytosol. Cell. Microbiol. 2019, 21, e13074. [Google Scholar] [CrossRef]
- Burress, H.; Kellner, A.; Guyette, J.; Tatulian, S.A.; Teter, K. HSC70 and HSP90 chaperones perform complementary roles in translocation of the cholera toxin A1 subunit from the endoplasmic reticulum to the cytosol. J. Biol. Chem. 2019, 294, 12122–12131. [Google Scholar] [CrossRef]
- Dever, T.; Kinzy, T.; Pavitt, G. Mechanism and Regulation of Protein Synthesis in Saccharomyces cerevisiae. Genetics 2016, 203, 65–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fieldhouse, R.J.; Jørgensen, R.; Lugo, M.R.; Merrill, A.R. The 1.8 A Cholix Toxin Crystal Structure in Complex with NAD+ and Evidence for a New Kinetic Model. J. Biol. Chem. 2012, 287, 21176–21188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wedekind, J.E.; Trame, C.B.; Dorywalska, M.; Koehl, P.; Raschke, T.M.; McKee, M.; Fitzgerald, D.; Collier, R.J.; McKay, D.B. Refined crystallographic structure of Pseudomonas aeruginosa exotoxin A and its implications for the molecular mechanism of toxicity 1 1Edited by D. Rees. J. Mol. Biol. 2001, 314, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.X.; London, E. Involvement of denaturation-like changes in Pseudomonas exotoxin a hydrophobicity and membrane penetration determined by characterization of pH and thermal transitions. Roles of two distinct conformationally altered states. J. Biol. Chem. 1990, 265, 8636–8641. [Google Scholar]
- Zheng, H.; Chordia, M.; Cooper, D.R.; Chruszcz, M.; Muller, P.; Sheldrick, G.M.; Minor, W. Validation of metal-binding sites in macromolecular structures with the CheckMyMetal web server. Nat. Protoc. 2013, 9, 156–170. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.C.; Efstratiou, A.; Mokrousov, I.; Mutreja, A.; Das, B.; Ramamurthy, T. Diphtheria. Nat. Rev. Dis. Prim. 2019, 5, 1–18. [Google Scholar] [CrossRef]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Establishment of Pseudomonas aeruginosa infection: Lessons from a versatile opportunist1*Address for correspondence: Channing Laboratory, 181 Longwood Avenue, Boston, MA 02115, USA. Microbes Infect. 2000, 2, 1051–1060. [Google Scholar] [CrossRef]
- Awasthi, S.P.; Asakura, M.; Chowdhury, N.; Neogi, S.B.; Hinenoya, A.; Golbar, H.M.; Yamate, J.; Arakawa, E.; Tada, T.; Ramamurthy, T.; et al. Novel Cholix Toxin Variants, ADP-Ribosylating Toxins in Vibrio cholerae Non-O1/Non-O139 Strains, and Their Pathogenicity. Infect. Immun. 2012, 81, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Purdy, A.; Rohwer, F.; Edwards, R.A.; Azam, F.; Bartlett, D.H. A Glimpse into the Expanded Genome Content of Vibrio cholerae through Identification of Genes Present in Environmental Strains. J. Bacteriol. 2005, 187, 2992–3001. [Google Scholar] [CrossRef] [Green Version]
- Romero, A.; Saraceni, P.R.; Merino, S.; Figueras, A.; Tomás, J.M.; Novoa, B. The Animal Model Determines the Results of Aeromonas Virulence Factors. Front. Microbiol. 2016, 7, 1245. [Google Scholar] [CrossRef] [Green Version]
- Ponnusamy, D.; Kozlova, E.V.; Sha, J.; Erova, T.E.; Azar, S.R.; Fitts, E.C.; Kirtley, M.L.; Tiner, B.L.; Andersson, J.A.; Grim, C.J.; et al. Cross-talk among flesh-eating Aeromonas hydrophila strains in mixed infection leading to necrotizing fasciitis. Proc. Natl. Acad. Sci. USA 2016, 113, 722–727. [Google Scholar] [CrossRef] [Green Version]
- Turgeon, Z.; Jørgensen, R.; Visschedyk, D.; Edwards, P.R.; Legree, S.; McGregor, C.; Fieldhouse, R.J.; Mangroo, D.; Schapira, M.; Merrill, A.R. Newly Discovered and Characterized Antivirulence Compounds Inhibit Bacterial Mono-ADP-Ribosyltransferase Toxins. Antimicrob. Agents Chemother. 2010, 55, 983–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallera, D.A.; Kreitman, R.J. Immunotoxins Targeting B Cell Malignancy—Progress and Problems with Immunogenicity. Biomedicines 2018, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarnovsky, R.; Tendler, T.; Makowski, M.; Kiley, M.; Antignani, A.; Traini, R.; Zhang, J.; Hassan, R.; Fitzgerald, D.J. Initial characterization of an immunotoxin constructed from domains II and III of cholera exotoxin. Cancer Immunol. Immunother. 2009, 59, 737–746. [Google Scholar] [CrossRef]
- Gildea, R.; Waterman, D.G.; Parkhurst, J.M.; Axford, D.; Sutton, G.; Stuart, D.; Sauter, N.K.; Evans, G.; Winter, G. New methods for indexing multi-lattice diffraction data. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 2652–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P. Scaling, and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Computational Project; Number the CCP4 suite: Programs for protein crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 1994, 50, 760–763. [CrossRef]
- McCoy, A.J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 63, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PE | Cholix | |||
|---|---|---|---|---|
| % ID | rmsd (Å) [Cα]1 | % ID | rmsd (Å) [Cα]1 | |
| Overall | 63.6 | 2.5 [593] | 35.4 | 2.9 [575] |
| domain Ia | 54.4 | 1.9 [239] | 33.0 | 2.3 [224] |
| domain II | 58.8 | 1.6 [111] | 33.6 | 2.3 [113] |
| domain III | 71.9 | 1.2 [209] | 45.8 | 1.9 [203] |
| AE (PDB 6Z5H) | |
|---|---|
| Data collection | |
| Beamline, λ (Å) | DLS-I03, 0.976 |
| Space group | P1211 |
| Cell dimensions | |
| a, b, c (Å) | 55.4, 140.2, 96.8 |
| α, β, γ (°) | 90.0, 101.8, 90.0 |
| Resolution (Å) | 2.3-56.4 (2.30-2.36)1 |
| No. total/unique reflections | 757,122/60,033 |
| Rmerge | 0.128 (0.845)1 |
| Rpim | 0.055 (0.414)1 |
| CC1/2 | 0.998 (0.895)1 |
| I/σI | 12.5 (2.8)1 |
| Completeness (%) | 99.0 (87.2)1 |
| Redundancy | 12.6 (9.7)1 |
| Refinement | |
| No. used reflections | 56,930 |
| Rwork/Rfree (%) | 20.4/24.7 |
| B-factors (Å2) | |
| Protein (all atoms)2 | 39.5/42.3 |
| Na+ ion2 | 29.0/26.9 |
| Water | 34.7 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 1.33 |
| Ramachandran statistics | |
| Favoured (%) | 96.5 |
| Outliers (%) | 0.00 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masuyer, G. Crystal Structure of Exotoxin A from Aeromonas Pathogenic Species. Toxins 2020, 12, 397. https://doi.org/10.3390/toxins12060397
Masuyer G. Crystal Structure of Exotoxin A from Aeromonas Pathogenic Species. Toxins. 2020; 12(6):397. https://doi.org/10.3390/toxins12060397
Chicago/Turabian StyleMasuyer, Geoffrey. 2020. "Crystal Structure of Exotoxin A from Aeromonas Pathogenic Species" Toxins 12, no. 6: 397. https://doi.org/10.3390/toxins12060397
APA StyleMasuyer, G. (2020). Crystal Structure of Exotoxin A from Aeromonas Pathogenic Species. Toxins, 12(6), 397. https://doi.org/10.3390/toxins12060397