Semiquantitation of Paralytic Shellfish Toxins by Hydrophilic Interaction Liquid Chromatography-Mass Spectrometry Using Relative Molar Response Factors

 ,
,
Abstract
:1. Introduction
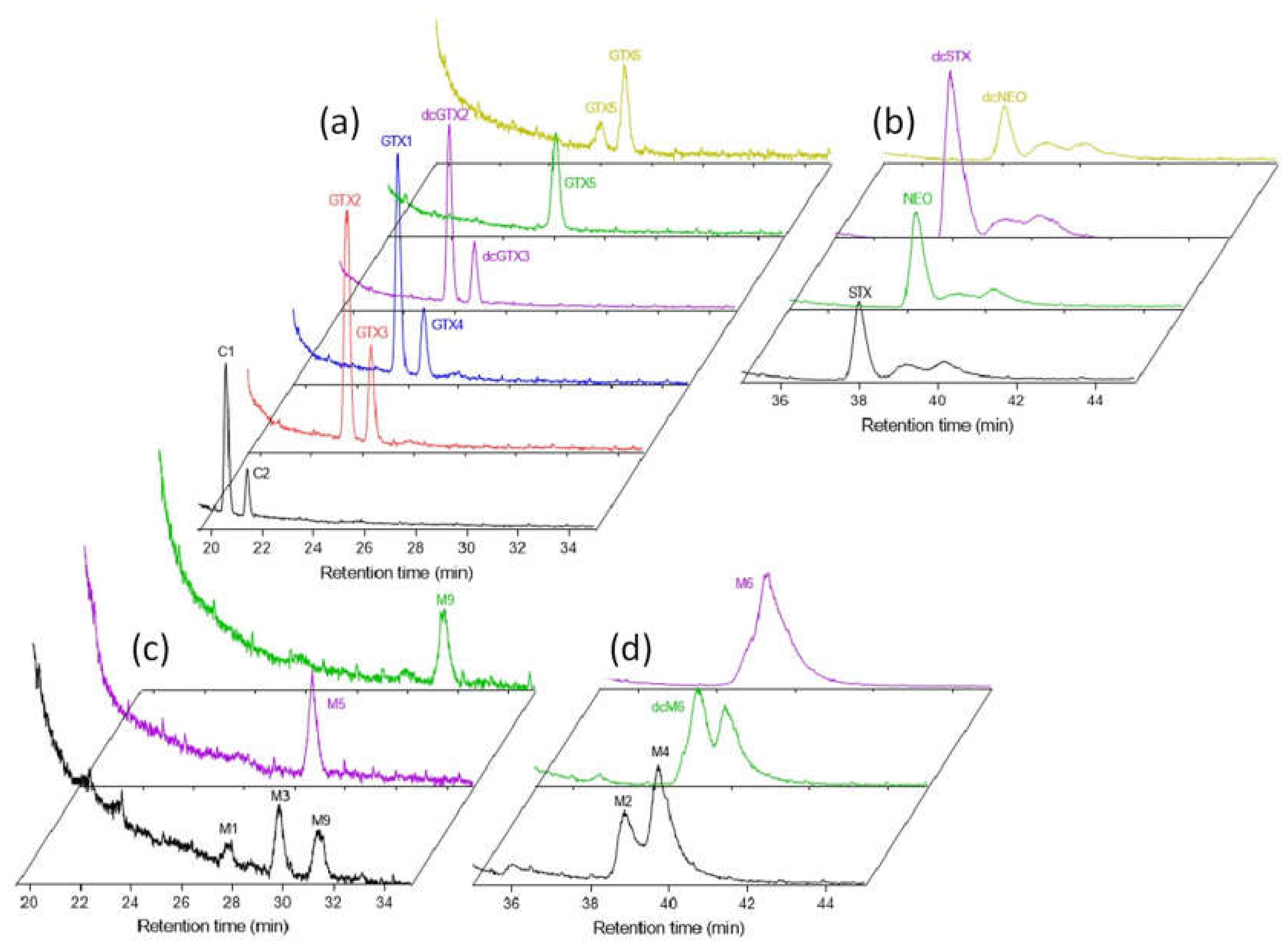
2. Results and Discussion
2.1. Semipurification and Quantitation of M-Toxins
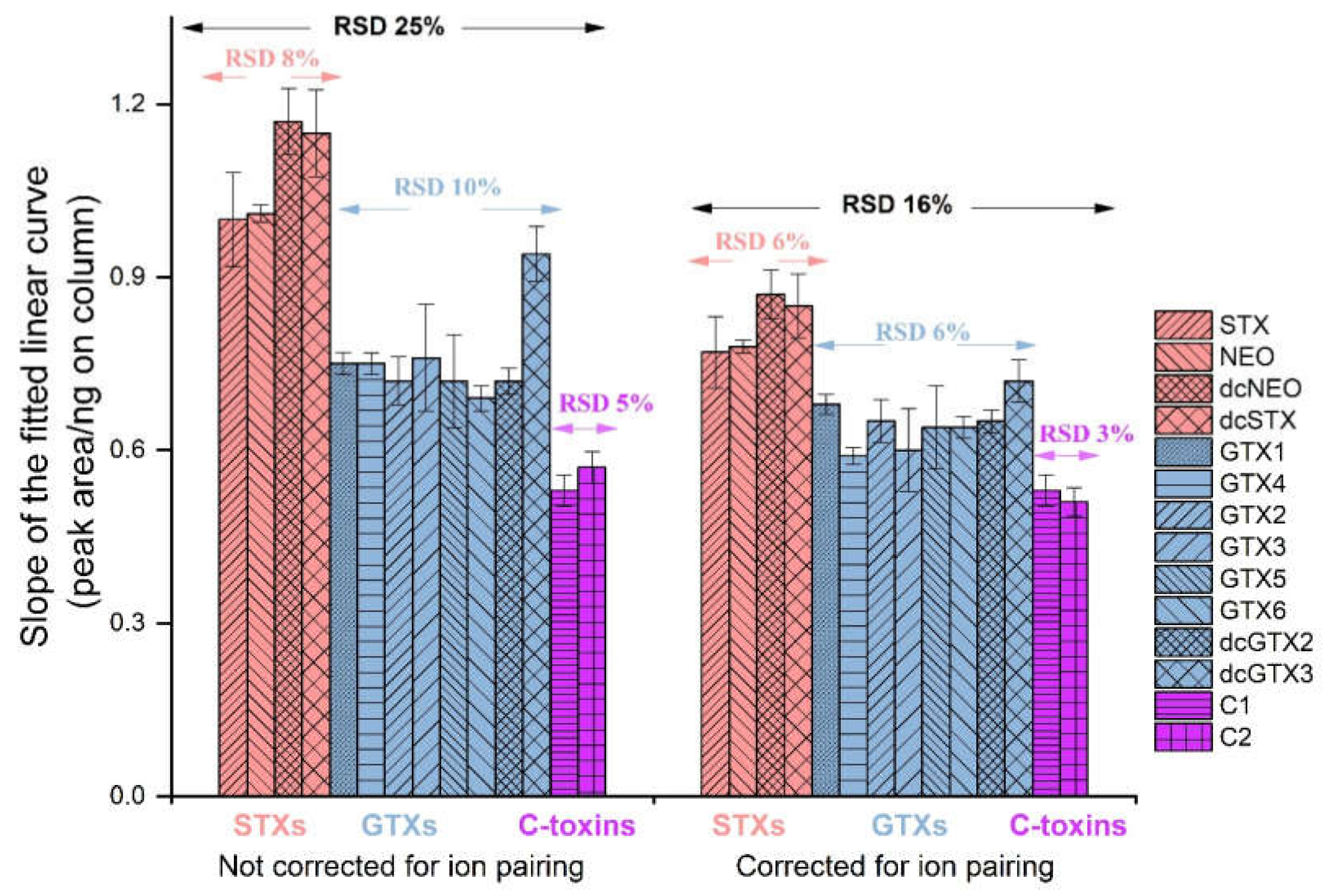
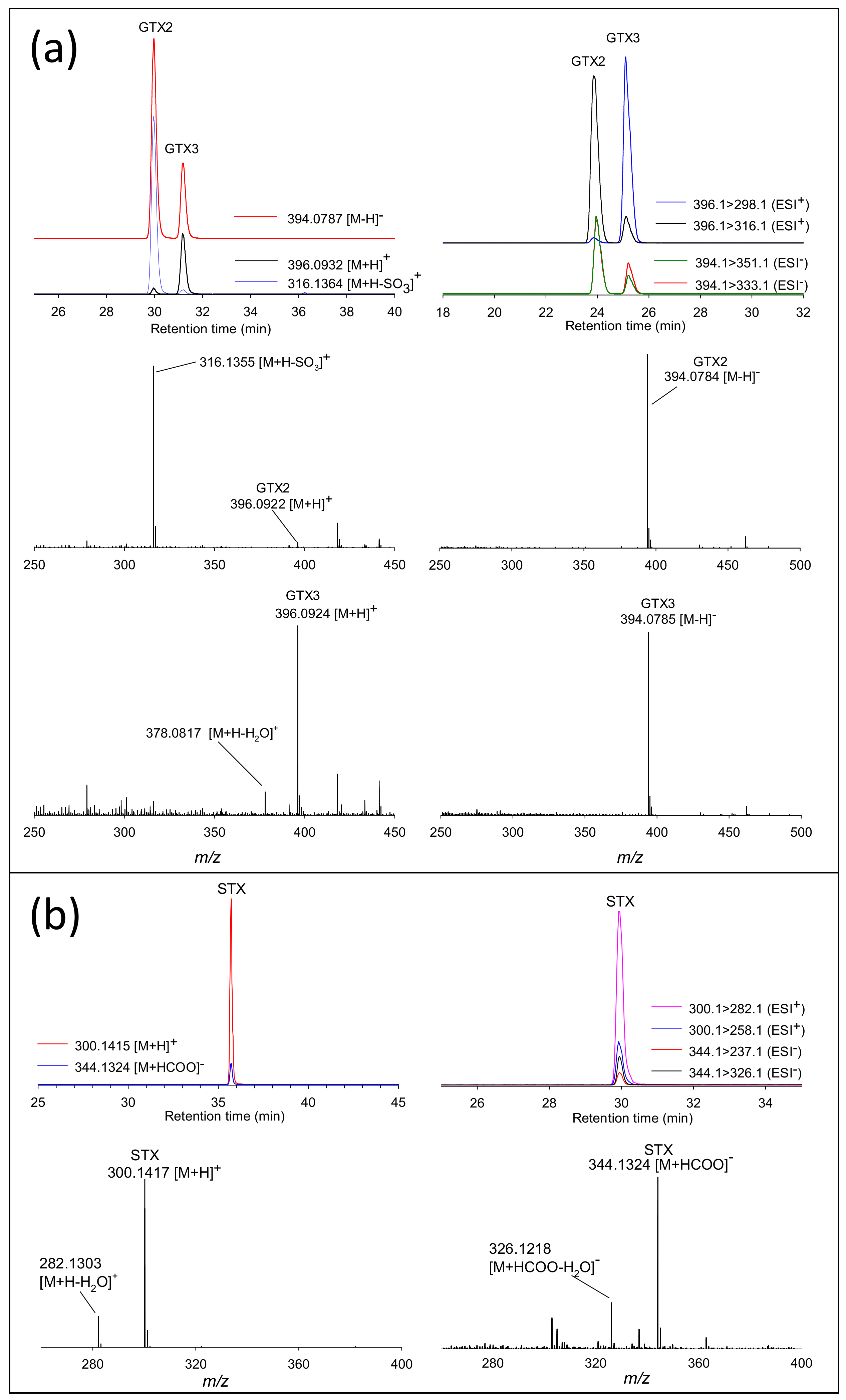
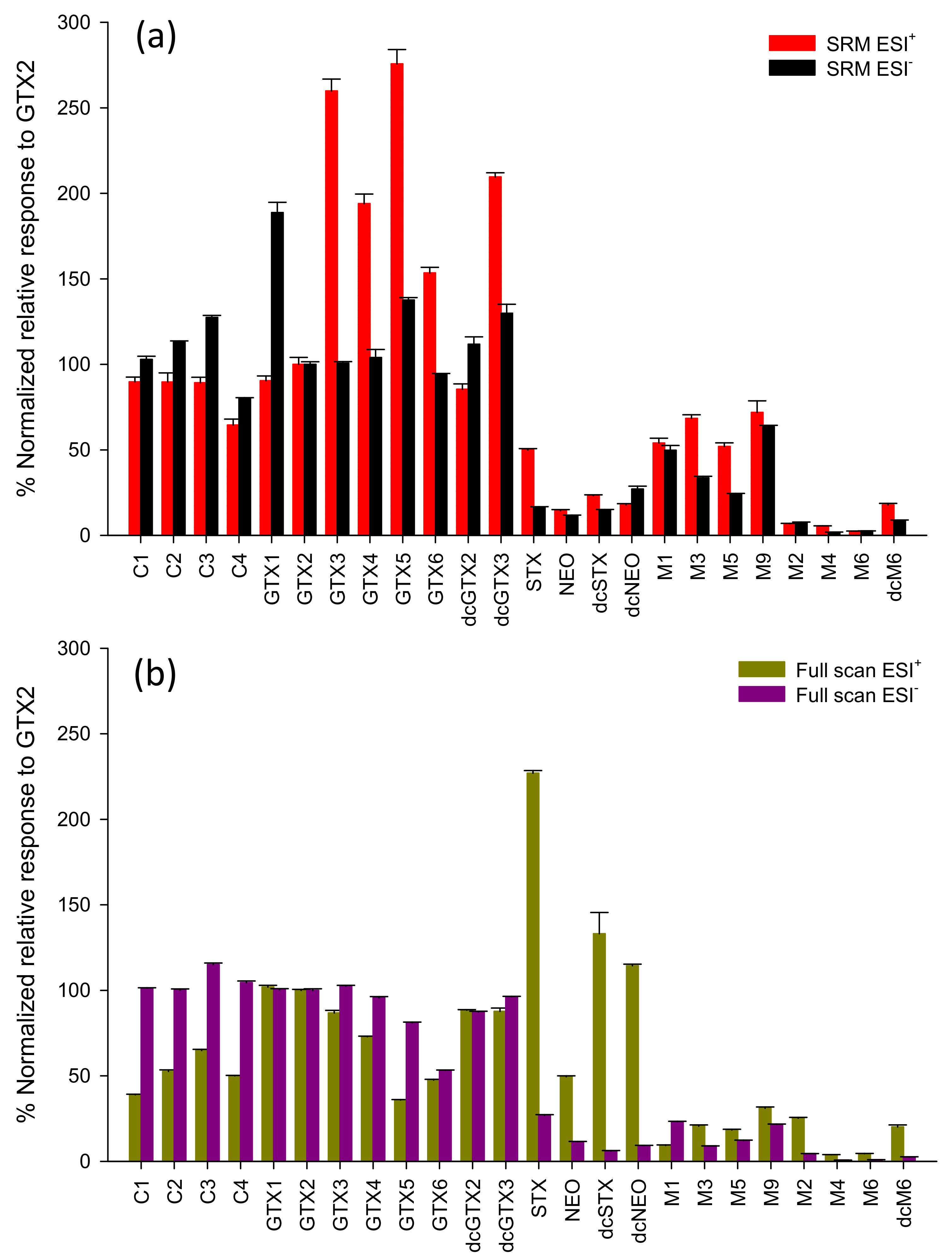
2.2. Evaluating RMRs for PSTs in LC-MS
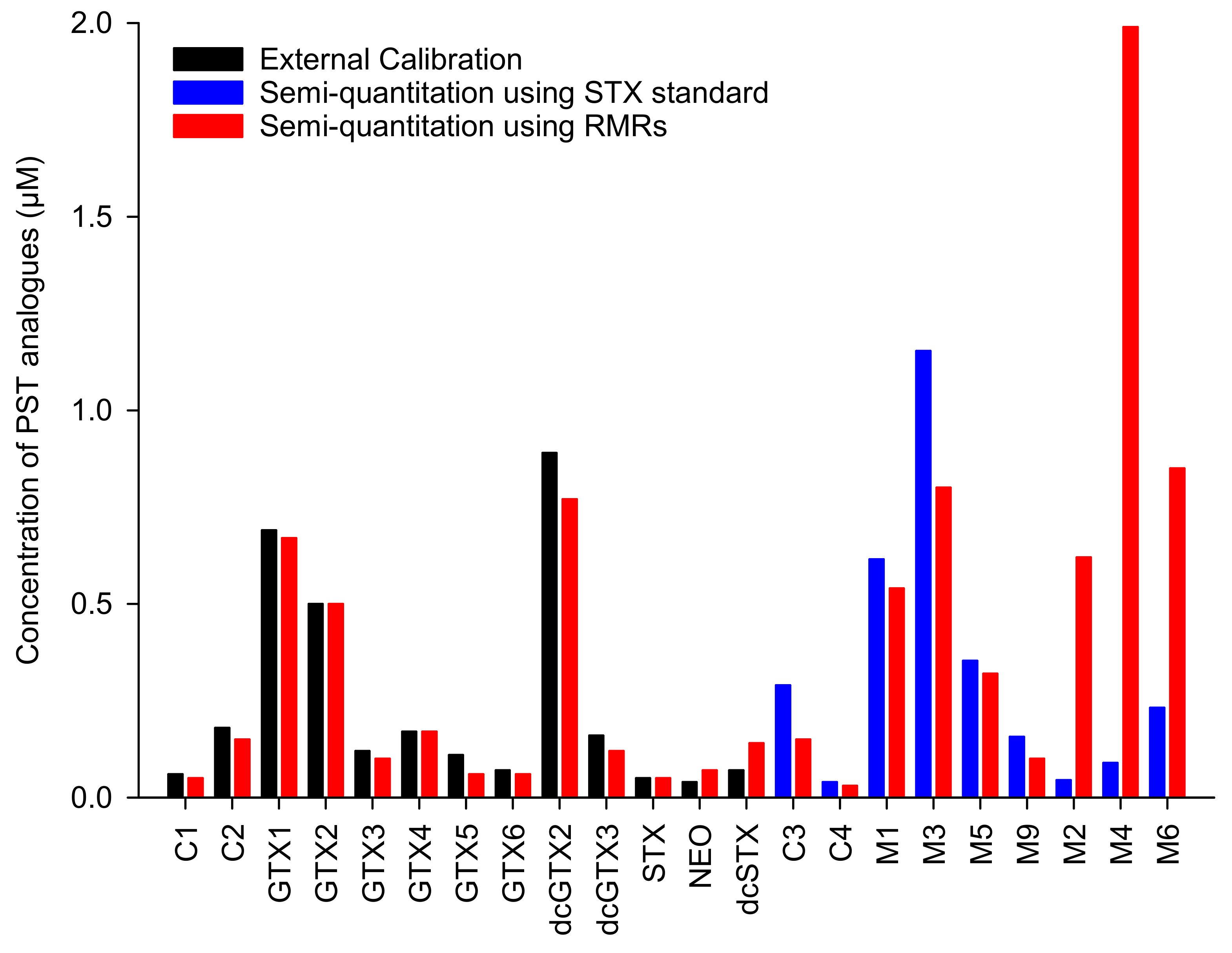
2.3. Semiquantitation of PSTs by LC-MS/MS Using RMRs
3. Conclusions
4. Materials and Methods
4.1. Standards and Chemicals
4.2. Semipurification of M-Toxins
4.3. Sample Preparation
4.4. LC-CAD-MS Analysis
4.5. LC-MS Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wiese, M.; D’Agostino, P.M.; Mihali, T.K.; Moffitt, M.C.; Neilan, B.A. Neurotoxic alkaloids: Saxitoxin and its analogs. Mar. Drugs 2010, 8, 2185–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etheridge, S.M. Paralytic shellfish poisoning: Seafood safety and human health perspectives. Toxicon 2010, 56, 108–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dell’Aversano, C.; Walter, J.A.; Burton, I.W.; Stirling, D.J.; Fattorusso, E.; Quilliam, M.A. Isolation and structure elucidation of new and unusual saxitoxin analogues from mussels. J. Nat. Prod. 2008, 71, 1518–1523. [Google Scholar] [CrossRef] [PubMed]
- Vale, P. Metabolites of saxitoxin analogues in bivalves contaminated by Gymnodinium catenatum. Toxicon 2010, 55, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Ma, J.; Cao, J.; Wang, Q.; Yu, R.; Thomas, K.; Quilliam, M.A. Analysis of paralytic shellfish toxins and their metabolites in shellfish from the North Yellow Sea of China. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2012, 29, 1455–1464. [Google Scholar] [CrossRef]
- Ding, L.; Qiu, J.; Li, A. Proposed biotransformation pathways for new metabolites of paralytic shellfish toxins based on field and experimental mussel samples. J. Agric. Food Chem. 2017, 65, 5494–5502. [Google Scholar] [CrossRef]
- Quilliam, M.A.; Li, A.; Lewis, N.; McCarron, P.; Thomas, K.; Walter, J.A. Biotransformation and chemical degradation of paralytic shellfish toxins in mussels. In Proceedings of the 17th International Conference on Harmful Algae, Florianópolis, Brazil, 9–14 October 2016; pp. 118–121. [Google Scholar]
- Qiu, J.; Meng, F.; Ding, L.; Che, Y.; McCarron, P.; Beach, D.G.; Li, A. Dynamics of paralytic shellfish toxins and their metabolites during timecourse exposure of scallops Chlamys farreri and mussels Mytilus galloprovincialis to Alexandrium pacificum. Aquat. Toxicol. 2018, 200, 233–240. [Google Scholar] [CrossRef]
- Qiu, J.; Rafuse, C.; Lewis, N.I.; Li, A.; Meng, F.; Beach, D.G.; McCarron, P. Screening of cyclic imine and paralytic shellfish toxins in isolates of the genus Alexandrium (Dinophyceae) from Atlantic Canada. Harmful Algae 2018, 77, 108–118. [Google Scholar] [CrossRef]
- D’Agostino, P.M.; Boundy, M.J.; Harwood, T.D.; Carmichael, W.W.; Neilan, B.A.; Wood, S.A. Re-evaluation of paralytic shellfish toxin profiles in cyanobacteria using hydrophilic interaction liquid chromatography-tandem mass spectrometry. Toxicon 2019, 158, 1–7. [Google Scholar] [CrossRef]
- Lawrence, J.F.; Menard, C.; Cleroux, C. Evaluation of prechromatographic oxidation for liquid chromatographic determination of paralytic shellfish poisons in shellfish. J. AOAC Int. 1995, 78, 514–520. [Google Scholar] [CrossRef]
- Rourke, W.A.; Murphy, C.J.; Pitcher, G.; van de Riet, J.M.; Burns, B.G.; Thomas, K.M.; Quilliam, M.A. Rapid postcolumn methodology for determination of paralytic shellfish toxins in shellfish tissue. J. AOAC Int. 2008, 91, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Turner, A.D.; Hatfield, R.G.; Rapkova-Dhanji, M.; Norton, D.M.; Algoet, M.; Lees, D.N. Single-laboratory validation of a refined AOAC HPLC method 2005.06 for oysters, cockles, and clams in UK Shellfish. J. AOAC Int. 2010, 93, 1482–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turrell, E.; Stobo, L.; Lacaze, J.P.; Piletsky, S.; Piletska, E. Optimization of hydrophilic interaction liquid chromatography/mass spectrometry and development of solid-phase extraction for the determination of paralytic shellfish poisoning toxins. J. AOAC Int. 2008, 91, 1372–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boundy, M.J.; Selwood, A.I.; Harwood, D.T.; McNabb, P.S.; Turner, A.D. Development of a sensitive and selective liquid chromatography–mass spectrometry method for high throughput analysis of paralytic shellfish toxins using graphitised carbon solid phase extraction. J. Chromatogr. A 2015, 1387, 1–12. [Google Scholar] [CrossRef]
- Thomas, K.; Beach, D.G.; Reeves, K.L.; Gibbs, R.; Kerrin, E.S.; McCarron, P.; Quilliam, M.A. Hydrophilic interaction liquid chromatography-tandem mass spectrometry for quantitation of paralytic shellfish toxins: Validation and application to reference materials. Anal. Bioanal. Chem. 2017, 409, 5675–5687. [Google Scholar] [CrossRef] [PubMed]
- Dell’Aversano, C.; Hess, P.; Quilliam, M.A. Hydrophilic interaction liquid chromatography-mass spectrometry for the analysis of paralytic shellfish poisoning (PSP) toxins. J. Chromatogr. A 2005, 1081, 190–201. [Google Scholar] [CrossRef]
- Turner, A.D.; McNabb, P.S.; Harwood, D.T.; Selwood, A.I.; Boundy, M.J. Single-Laboratory validation of a multitoxin ultra-performance LC-Hydrophilic Interaction LC-MS/MS method for quantitation of paralytic shellfish toxins in bivalve shellfish. J. AOAC Int. 2015, 98, 609–621. [Google Scholar] [CrossRef]
- Zhuo, L.; Yin, Y.; Fu, W.; Qiu, B.; Lin, Z.; Yang, Y.; Zheng, L.; Li, J.; Chen, G. Determination of paralytic shellfish poisoning toxins by HILIC-MS/MS coupled with dispersive solid phase extraction. Food Chem. 2013, 137, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Blay, P.; Hui, J.P.M.; Chang, J.; Melanson, J.E. Screening for multiple classes of marine biotoxins by liquid chromatography-high-resolution mass spectrometry. Anal. Bioanal. Chem. 2011, 400, 577–585. [Google Scholar] [CrossRef]
- Leito, I.; Herodes, K.; Huopolainen, M.; Virro, K.; Kunnapas, A.; Kruve, A.; Tanner, R. Towards the electrospray ionization mass spectrometry ionization efficiency scale of organic compounds. Rapid Commun. Mass Spectrom. 2008, 22, 379–384. [Google Scholar] [CrossRef]
- Otero, P.; Rodrigues, P. Marker compounds, relative response factors, and toxic equivalent factors. In Seafood and Freshwater Toxins: Pharmacology, Physiology, and Detection, 3rd ed.; Botana, L.M., Ed.; CRC Press: Boca Raton, FL, USA, 2014; pp. 57–76. [Google Scholar]
- Kilcoyne, J.; Nulty, C.; Jauffrais, T.; McCarron, P.; Herve, F.; Foley, B.; Rise, F.; Crain, S.; Wilkins, A.L.; Twiner, M.J.; et al. Isolation, structure elucidation, relative LC-MS response, and in vitro toxicity of azaspiracids from the dinoflagellate Azadinium spinosum. J. Nat. Prod. 2014, 77, 2465–2474. [Google Scholar] [CrossRef] [PubMed]
- Kilcoyne, J.; McCarron, P.; Twiner, M.J.; Nulty, C.; Crain, S.; Quilliam, M.A.; Rise, F.; Wilkins, A.L.; Miles, C.O. Epimers of azaspiracids: Isolation, structural elucidation, relative LC-MS response, and in vitro toxicity of 37-epi-azaspiracid-1. Chem. Res. Toxicol. 2014, 27, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Kilcoyne, J.; Twiner, M.J.; McCarron, P.; Crain, S.; Giddings, S.D.; Foley, B.; Rise, F.; Hess, P.; Wilkins, A.L.; Miles, C.O. Structure elucidation, relative LC-MS response and in vitro toxicity of azaspiracids 7–10 isolated from mussels (Mytilus edulis). J. Agric. Food Chem. 2015, 63, 5083–5091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zendong, Z.; Sibat, M.; Herrenknecht, C.; Hess, P.; McCarron, P. Relative molar response of lipophilic marine algal toxins in liquid chromatography/electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2017, 31, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Yogi, K.; Oshiro, N.; Inafuku, Y.; Hirama, M.; Yasumoto, T. Detailed LC-MS/MS analysis of ciguatoxins revealing distinct regional and species characteristics in fish and causative alga from the pacific. Anal. Chem. 2011, 83, 8886–8891. [Google Scholar] [CrossRef]
- Burton, I.W.; Quilliam, M.A.; Walter, J.A. Quantitative 1H NMR with external standards: Use in preparation of calibration solutions for algal toxins and other natural products. Anal. Chem. 2005, 77, 3123–3131. [Google Scholar] [CrossRef]
- Perez, R.A.; Rehmann, N.; Crain, S.; LeBlanc, P.; Craft, C.; MacKinnon, S.; Reeves, K.; Burton, I.W.; Walter, J.A.; Hess, P.; et al. The preparation of certified calibration solutions for azaspiracid-1, -2, and -3, potent marine biotoxins found in shellfish. Anal. Bioanal. Chem. 2010, 398, 2243–2252. [Google Scholar] [CrossRef] [Green Version]
- Beach, D.G.; Crain, S.; Lewis, N.; LeBlanc, P.; Hardstaff, W.R.; Perez, R.A.; Giddings, S.D.; Martinez-Farina, C.F.; Stefanova, R.; Burton, I.W.; et al. Development of certified reference materials for diarrhetic shellfish poisoning toxins, part 1: Calibration solutions. J. AOAC Int. 2016, 99, 1151–1162. [Google Scholar] [CrossRef]
- Thomas, K.; Wechsler, D.; Chen, Y.M.; Crain, S.; Quilliam, M.A. Analysis of natural toxins by liquid chromatography-chemiluminescence nitrogen detection and application to the preparation of certified reference materials. J. AOAC Int. 2016, 99, 1173–1184. [Google Scholar] [CrossRef]
- Megoulas, N.C.; Koupparis, M.A. Twenty years of evaporative light scattering detection. Crit. Rev. Anal. Chem. 2005, 35, 301–316. [Google Scholar] [CrossRef]
- Sinclair, I.; Gallagher, R. Charged Aerosol Detection: Factors for consideration in its use as a generic quantitative detector. Chromatogr. Today 2008, 1, 5–9. Available online: https://www.chromatographytoday.com/article/autosamplers/36/chromatography-today-news/charged-aerosol-detection-factors-for-consideration-in-its-use-as-a-generic-quantitative-detector/524/download (accessed on 13 June 2020).
- Hutchinson, J.P.; Li, J.; Farrell, W.; Groeber, E.; Szucs, R.; Dicinoski, G.; Haddad, P.R. Universal response model for a corona charged aerosol detector. J. Chromatogr. A 2010, 1217, 7418–7427. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.J.; Heaton, J.C.; Underwood, T.; Boughtflower, R.; McCalley, D.V. Performance of charged aerosol detection with hydrophilic interaction chromatography. J. Chromatogr. A 2015, 1405, 72–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, T.; Oshima, Y.; Yasumoto, T. Structures of two paralytic shellfish toxins, gonyautoxins V and VI, isolated from a tropical dinoflagellate Pyrodinium bahamense var. compressa. Agric. Biol. Chem. 1982, 46, 1861–1864. [Google Scholar]
- Kodama, M.; Ogata, T.; Noguchi, T.; Maruyama, J.; Hashimoto, K. Occurrence of saxitoxin and other toxins in the liver of the pufferfish Takifugu pardalis. Toxicon 1983, 21, 897–900. [Google Scholar] [CrossRef]
- Ito, K.; Asakawa, M.; Sida, Y.; Miyazawa, K. Occurrence of paralytic shellfish poison (psp) in the starfish asterina pectinifera collected from the kure bay, hiroshima prefecture, japan. Toxicon 2003, 41, 291–295. [Google Scholar] [CrossRef]
- Laycock, M.V.; Thibault, P.; Ayer, S.W.; Walter, J.A. Isolation and purification procedures for the preparation of paralytic shellfish poisoning toxin standards. Nat. Toxins 1994, 2, 175–183. [Google Scholar] [CrossRef]
- Beach, D.G.; Kerrin, E.S.; Thomas, K.; Quilliam, M.A.; McCarron, P. Capillary electrophoresis-tandem mass spectrometry for multiclass analysis of polar marine toxins. Anal. Bioanal. Chem. 2018, 410, 5405–5420. [Google Scholar] [CrossRef]
- Dörr, F.A.; Kovačević, B.; Maksić, Z.B.; Pinto, E.; Volmer, D.A. Intriguing differences in the gas-phase dissociation behavior of protonated and deprotonated gonyautoxin epimers. J. Am. Soc. Mass Spectrom. 2011, 22, 2011–2020. [Google Scholar] [CrossRef] [Green Version]
- Górecki, T.; Lynen, F.; Szucs, R.; Sandra, P. Universal response in liquid chromatography using charged aerosol detection. Anal. Chem. 2006, 78, 3186–3192. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin | LC-MS/MS | LC-HRMS | ||
|---|---|---|---|---|
| ESI+ | ESI− | ESI+ | ESI− | |
| C1 | 0.90 (3.3) | 1.0 (1.9) | 0.39 (0.8) | 1.0 (0.2) |
| C2 | 0.90 (5.6) | 1.1 (0.5) | 0.53 (1.7) | 1.0 (0.7) |
| C3 | 0.89 (3.3) | 1.3 (1.8) | 0.65 (0.9) | 1.2 (0.8) |
| C4 | 0.65 (4.6) | 0.80 (0.1) | 0.50 (0.6) | 1.1 (1.0) |
| GTX1 | 0.90 (3.3) | 1.9 (3.2) | 1.0 (1.0) | 1.0 (0.7) |
| GTX2 | 1.0 (4.0) | 1.0 (2.0) | 1.0 (0.6) | 1.0 (0.9) |
| GTX3 | 2.6 (2.7) | 1.0 (0.9) | 0.87 (2.3) | 1.0 (0.4) |
| GTX4 | 1.9 (3.1) | 1.0 (4.8) | 0.73 (0.5) | 0.96 (0.7) |
| GTX5 | 2.8 (2.9) | 1.8 (0.7) | 0.36 (0.8) | 0.81 (0.5) |
| GTX6 | 1.5 (2.0) | 0.94 (0.7) | 0.48 (0.8) | 0.53 (0.4) |
| dcGTX2 | 0.85 (3.5) | 1.1 (3.6) | 0.88 (0.5) | 0.87 (0.5) |
| dcGTX3 | 2.1 (1.4) | 1.3 (3.8) | 0.88 (2.3) | 0.96 (0.2) |
| STX | 0.5 (2.0) | 0.16 (3.7) | 2.2 (0.9) | 0.27 (1.5) |
| NEO | 0.14 (4.9) | 0.12 (3.5) | 0.50 (1.0) | 0.12 (1.7) |
| dcSTX | 0.23 (2.2) | 0.15 (1.3) | 1.3 (7.7) | 0.060 (5.0) |
| dcNEO | 0.18 (5.1) | 0.27 (7.4) | 1.1 (0.8) | 0.093 (0.8) |
| M1 | 0.54 (5.6) | 0.50 (6.0) | 0.093 (4.3) | 0.23 (0.9) |
| M3 | 0.68 (2.9) | 0.34 (2.9) | 0.21 (3.4) | 0.090 (0.1) |
| M5 | 0.52 (3.9) | 0.24 (1.7) | 0.18 (2.2) | 0.12 (1.6) |
| M9 | 0.72 (9.7) | 0.64 (1.3) | 0.31 (2.6) | 0.22 (0.5) |
| M2 | 0.069 (2.9) | 0.077 (1.3) | 0.25 (2.0) | 0.045 (0.9) |
| M4 | 0.055 (1.8) | 0.018 (5.6) | 0.039 (1.3) | 0.0071 (1.4) |
| M6 | 0.025 (4.0) | 0.026 (0.8) | 0.045 (4.4) | 0.010 (2.0) |
| dcM6 | 0.18 (5.0) | 0.086 (4.7) | 0.20 (5.0) | 0.025 (4.0) |
| Toxin | LC-HRMS | LC-MS/MS | |||||
|---|---|---|---|---|---|---|---|
| Ion | Exact m/z | Precursor (m/z) | Product (m/z) | DP (V) | CE (V) | ||
| C1/2 | [M+H-SO3]+ | 396.0932 | [M+NH4]+ | 493.1 | 316.1, 298.1 | 10, 10 | 35, 40 |
| C3/4 | [M+H-SO3]+ | 412.0881 | [M+NH4]+ | 509.1 | 332.1, 314.1 | 10, 10 | 35, 35 |
| GTX1 | [M+H-SO3]+ | 332.1313 | [M+H]+ | 412.1 | 332.1, 314.1 | 20, 120 | 20, 25 |
| GTX4 | [M+H]+ | 412.0881 | |||||
| GTX2 | [M+H-SO3]+ | 316.1364 | [M+H]+ | 396.1 | 316.1, 298.1 | 20, 120 | 20, 25 |
| GTX3 | [M+H]+ | 396.0932 | |||||
| dcGTX1 | [M+H-SO3]+ | 289.1255 | [M+H]+ | 369.1 | 289.1, 271.1 | 10, 120 | 20, 25 |
| dcGTX4 | [M+H]+ | 369.0823 | |||||
| dcGTX2 | [M+H-SO3]+ | 273.1306 | [M+H]+ | 353.1 | 273.1, 255.1 | 10, 120 | 20, 25 |
| dcGTX3 | [M+H]+ | 353.0874 | |||||
| GTX5 | [M+H]+ | 380.0983 | [M+H]+ | 380.1 | 300.1, 282.1 | 100, 100 | 20, 25 |
| GTX6 | [M+H]+ | 396.0932 | [M+H]+ | 396.1 | 316.1, 263.1 | 100, 100 | 20, 40 |
| STX | [M+H]+ | 300.1415 | [M+H]+ | 300.1 | 258.1, 282.1 | 140, 140 | 30, 25 |
| NEO | [M+H]+ | 316.1364 | [M+H]+ | 316.1 | 220.1, 298.1 | 160, 160 | 30, 25 |
| dcSTX | [M+H]+ | 257.1357 | [M+H]+ | 257.1 | 180.1, 222.1 | 160, 160 | 30, 25 |
| dcNEO | [M+H]+ | 273.1306 | [M+H]+ | 273.1 | 255.1, 126.1 | 160, 160 | 25, 45 |
| M1 | [M+H]+ | 396.0932 | [M+H]+ | 396.1 | 316.1, 148.1 | 100, 100 | 20, 40 |
| M3 | [M+H]+ | 412.0881 | [M+H]+ | 412.1 | 332.1, 235.1 | 120, 120 | 20, 40 |
| M5 | [M+H-H2O]+ | 396.0932 | [M+H]+ | 396.1 | 316.1, 239.1 | 120, 120 | 20, 40 |
| M7 | [M+H]+ | 412.0881 | [M+H]+ | 412.1 | 332.1, 314.1 | 120, 120 | 20, 25 |
| M9 | [M+H]+ | 428.0830 | [M+H]+ | 428.1 | 348.1, 330.1 | 100, 100 | 20, 20 |
| M2 | [M+H]+ | 316.1364 | [M+H]+ | 316.1 | 298.1, 148.1 | 100, 100 | 20, 40 |
| M4/M8 | [M+H]+ | 332.1313 | [M+H]+ | 332.1 | 314.1, 235.1 | 110, 110 | 20, 40 |
| M6 | [M+H-H2O]+ | 316.1364 | [M+H]+ | 316.1 | 257.1, 239.1 | 110, 110 | 20, 30 |
| M10 | [M+H]+ | 348.1262 | [M+H]+ | 348.1 | 330.1, 136.1 | 100, 100 | 30, 40 |
| dcM2 | [M+H]+ | 273.1306 | [M+H]+ | 273.1 | 255.1 | 80 | 20 |
| dcM4/8 | [M+H]+ | 289.1255 | [M+H]+ | 289.1 | 271.1 | 100 | 20 |
| dcM6 | [M+H-H2O]+ | 273.1306 | [M+H]+ | 273.1 | 214.1, 196.1 | 110, 110 | 30, 40 |
| dcM10 | [M+H]+ | 305.1204 | [M+H]+ | 305.1 | 287.1 | 140 | 20 |
| Toxin | Ion | LC-HRMS | LC-MS/MS | |||
|---|---|---|---|---|---|---|
| Exact m/z | Precursor (m/z) | Product (m/z) | DP (V) | CE (V) | ||
| C1/2 | [M-H]− | 474.0355 | 474.1 | 122.1, 456.1 | 70, 70 | 30, 20 |
| C3/4 | [M-H]− | 490.0304 | 490.1 | 410.1, 122.1 | 80, 80 | 30, 40 |
| GTX1/4 | [M-H]− | 410.0736 | 410.1 | 367.1, 349.1 | 100, 70 | 20, 30 |
| GTX2/3 | [M-H]− | 394.0787 | 394.1 | 351.1, 333.1 | 80, 100 | 25, 30 |
| dcGTX1/4 | [M-H]− | 367.0678 | 367.1 | 193.1, 349.1 | 100, 100 | 20, 20 |
| dcGTX2/3 | [M-H]− | 351.0728 | 351.1 | 333.1, 164.1 | 100, 100 | 25, 35 |
| GTX5 | [M-H]− | 378.0837 | 378.1 | 122.1, 360.1 | 70, 70 | 40, 25 |
| GTX6 | [M-H]− | 394.0787 | 394.1 | 376.1 | 60, 60 | 20 |
| STX | [M+HCOO]− | 344.1324 | 344.1 | 326.1, 237.1 | 70, 70 | 10, 20 |
| NEO | [M+HCOO]− | 360.1273 | 360.1 | 178.1, 342.1 | 80, 70 | 25, 10 |
| dcSTX | [M+HCOO]− | 301.1265 | 301.1 | 237.1, 136.1 | 80, 80 | 20, 30 |
| dcNEO | [M+HCOO]− | 317.1215 | 317.1 | 178.1, 124.1 | 60, 70 | 25, 40 |
| M1 | [M-H]− | 394.0787 | 394.1 | 122.1, 376.1 | 80, 60 | 40, 20 |
| M5 | [M-H-H2O]− | 394.0787 | 394.1 | 122.1, 376.1 | 80, 60 | 40, 20 |
| M3/7 | [M-H]− | 410.0736 | 410.1 | 374.1, 122.1 | 100, 100 | 20, 40 |
| M9 | [M-H]− | 426.0685 | 426.1 | 390.1, 122.1 | 80, 80 | 25, 40 |
| M2 | [M+HCOO]− | 360.1273 | 360.1 | 253.1, 164.1 | 100, 100 | 30, 40 |
| M4/M8 | [M+HCOO]− | 376.1222 | 376.1 | 294.1, 251.1 | 100, 100 | 20, 30 |
| M6 | [M+HCOO-H2O]− | 360.1273 | 360.1 | 253.1, 164.1 | 80, 80 | 25, 40 |
| M10 | [M+HCOO]− | 392.1171 | 392.1 | 374.1, 346.1 | 100, 100 | 20, 20 |
| dcM2 | [M+HCOO]− | 317.1215 | 317.1 | 299.1 | 100 | 20 |
| dcM4/8 | [M+HCOO]− | 333.1164 | 333.1 | 315.1 | 100 | 20 |
| dcM6 | [M+HCOO-H2O]− | 317.1215 | 317.1 | 253.1, 164.1 | 100, 100 | 30, 40 |
| dcM10 | [M+HCOO]− | 349.1113 | 349.1 | 331.1 | 100 | 20 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, J.; Wright, E.J.; Thomas, K.; Li, A.; McCarron, P.; Beach, D.G. Semiquantitation of Paralytic Shellfish Toxins by Hydrophilic Interaction Liquid Chromatography-Mass Spectrometry Using Relative Molar Response Factors. Toxins 2020, 12, 398. https://doi.org/10.3390/toxins12060398
Qiu J, Wright EJ, Thomas K, Li A, McCarron P, Beach DG. Semiquantitation of Paralytic Shellfish Toxins by Hydrophilic Interaction Liquid Chromatography-Mass Spectrometry Using Relative Molar Response Factors. Toxins. 2020; 12(6):398. https://doi.org/10.3390/toxins12060398
Chicago/Turabian StyleQiu, Jiangbing, Elliott J. Wright, Krista Thomas, Aifeng Li, Pearse McCarron, and Daniel G. Beach. 2020. "Semiquantitation of Paralytic Shellfish Toxins by Hydrophilic Interaction Liquid Chromatography-Mass Spectrometry Using Relative Molar Response Factors" Toxins 12, no. 6: 398. https://doi.org/10.3390/toxins12060398
APA StyleQiu, J., Wright, E. J., Thomas, K., Li, A., McCarron, P., & Beach, D. G. (2020). Semiquantitation of Paralytic Shellfish Toxins by Hydrophilic Interaction Liquid Chromatography-Mass Spectrometry Using Relative Molar Response Factors. Toxins, 12(6), 398. https://doi.org/10.3390/toxins12060398