A Role of Newly Found Auxiliary Site in Phospholipase A1 from Thai Banded Tiger Wasp (Vespa affinis) in Its Enzymatic Enhancement: In Silico Homology Modeling and Molecular Dynamics Insights

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
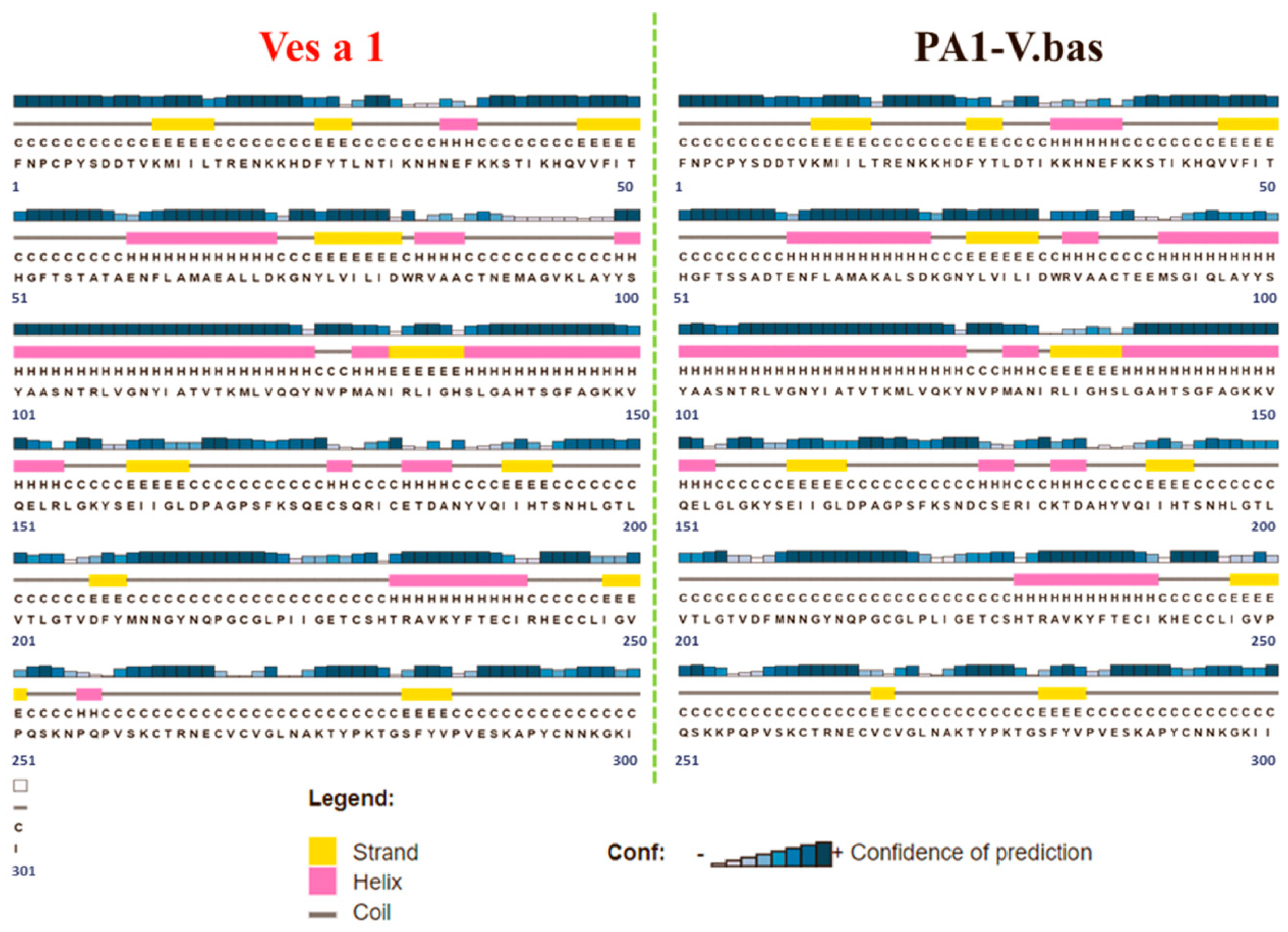
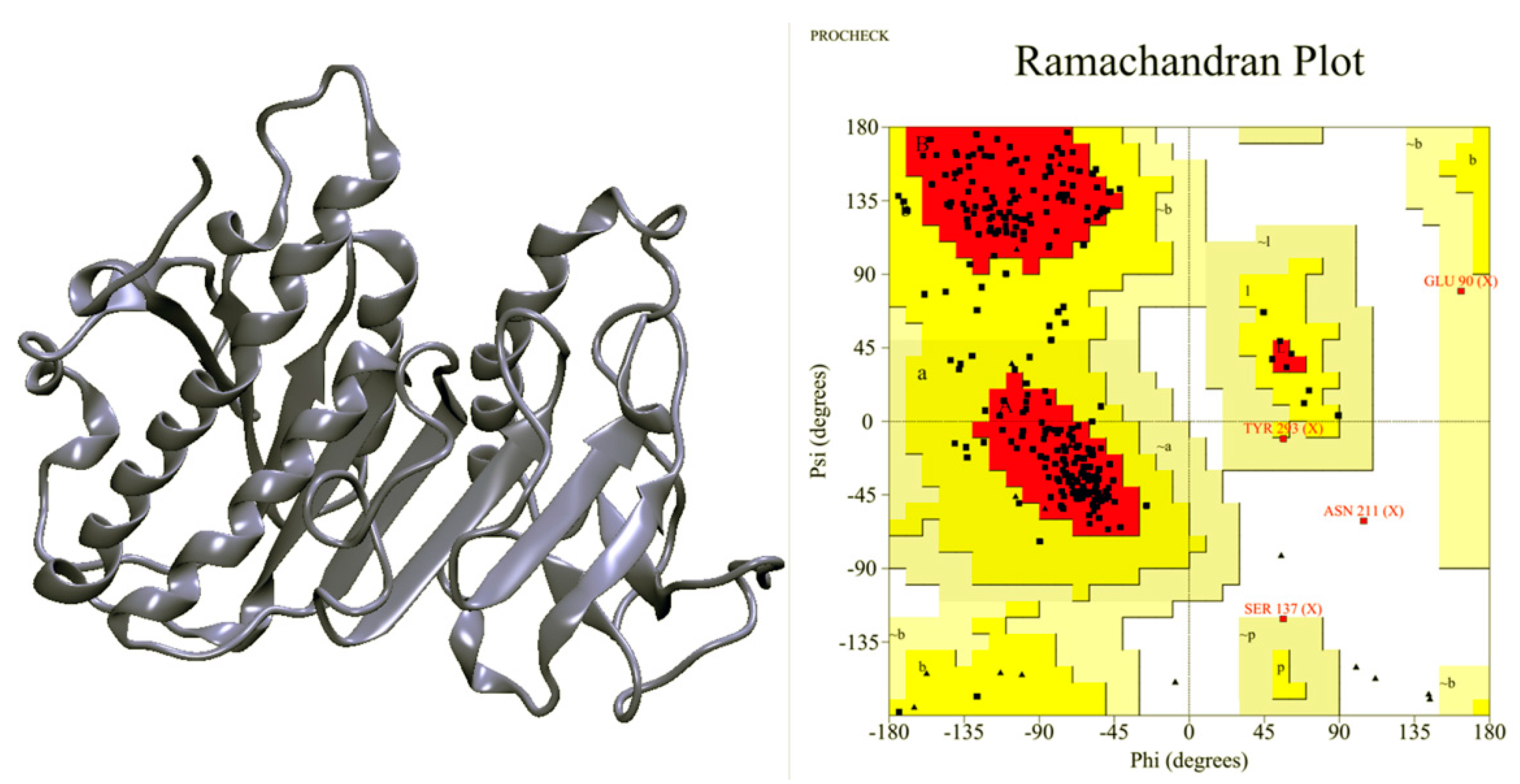
2.1. Ves a 1 Modeling
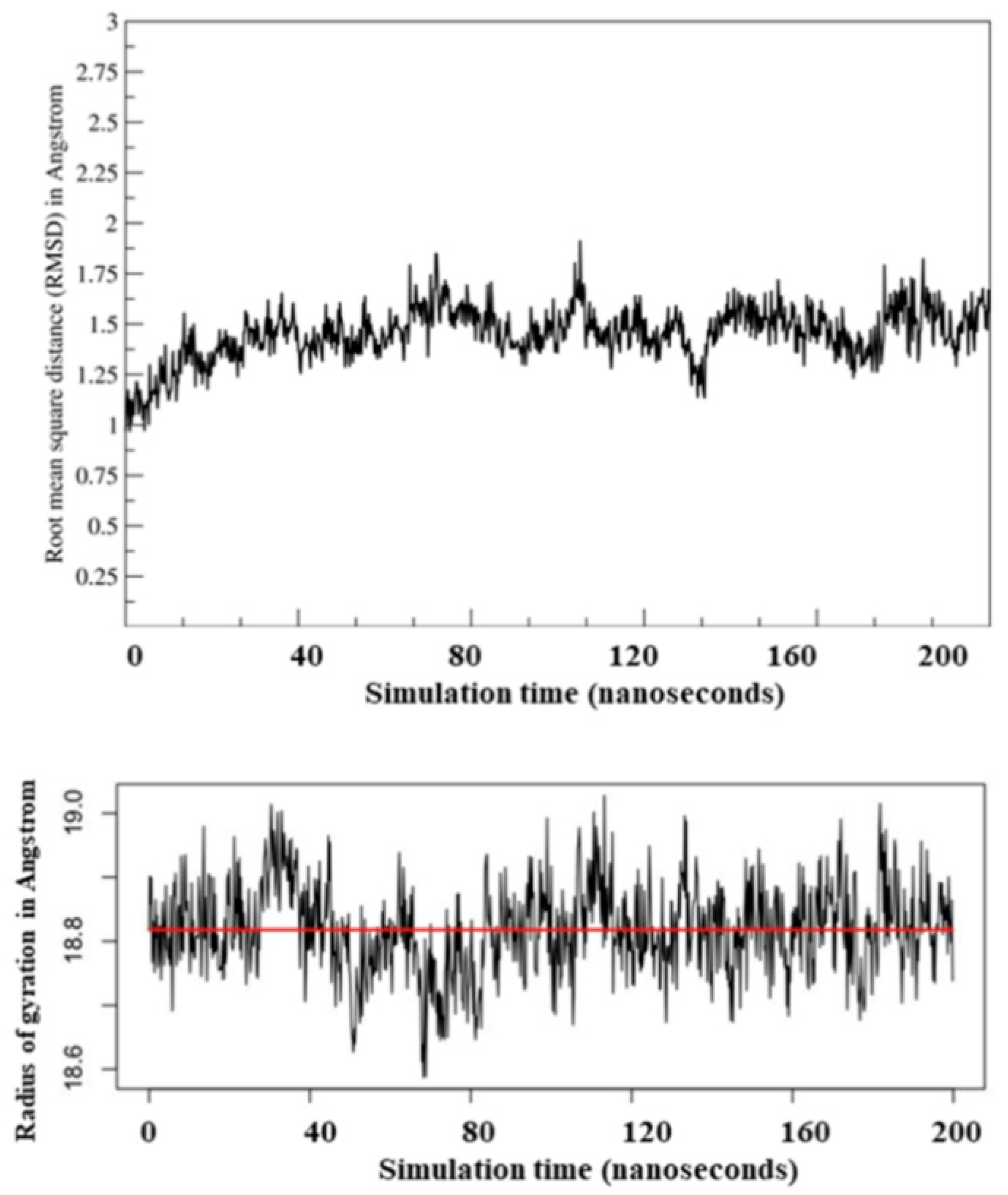
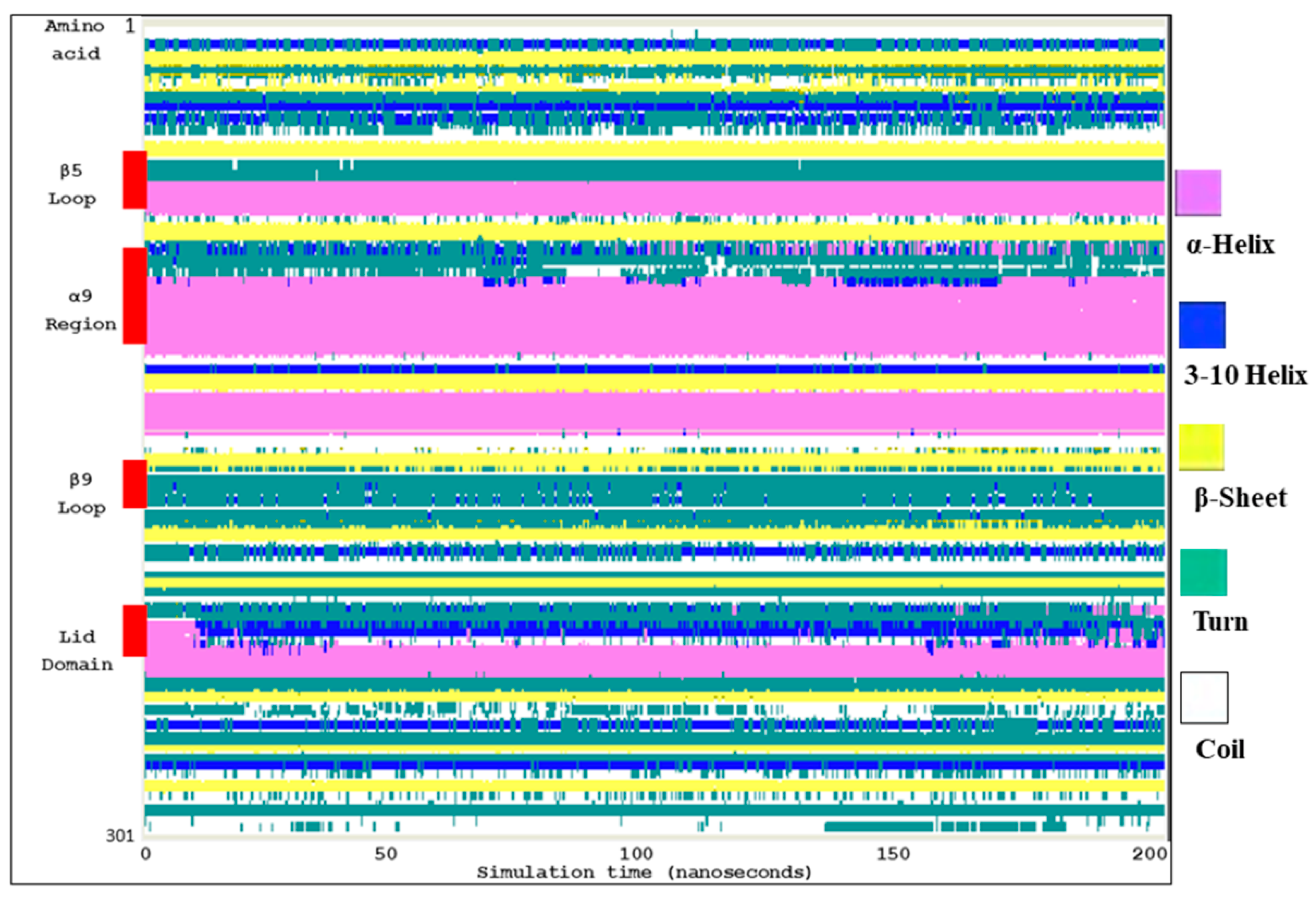
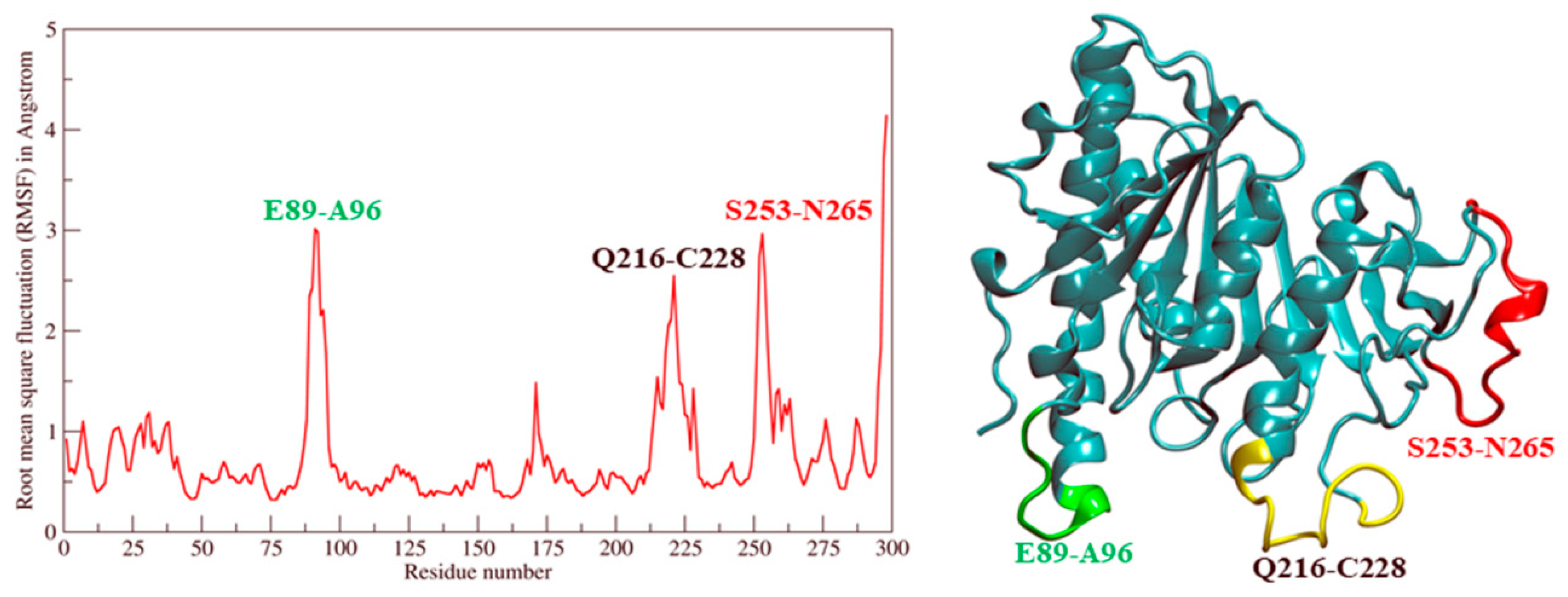
2.2. Molecular Dynamic Simulation and Molecular Docking
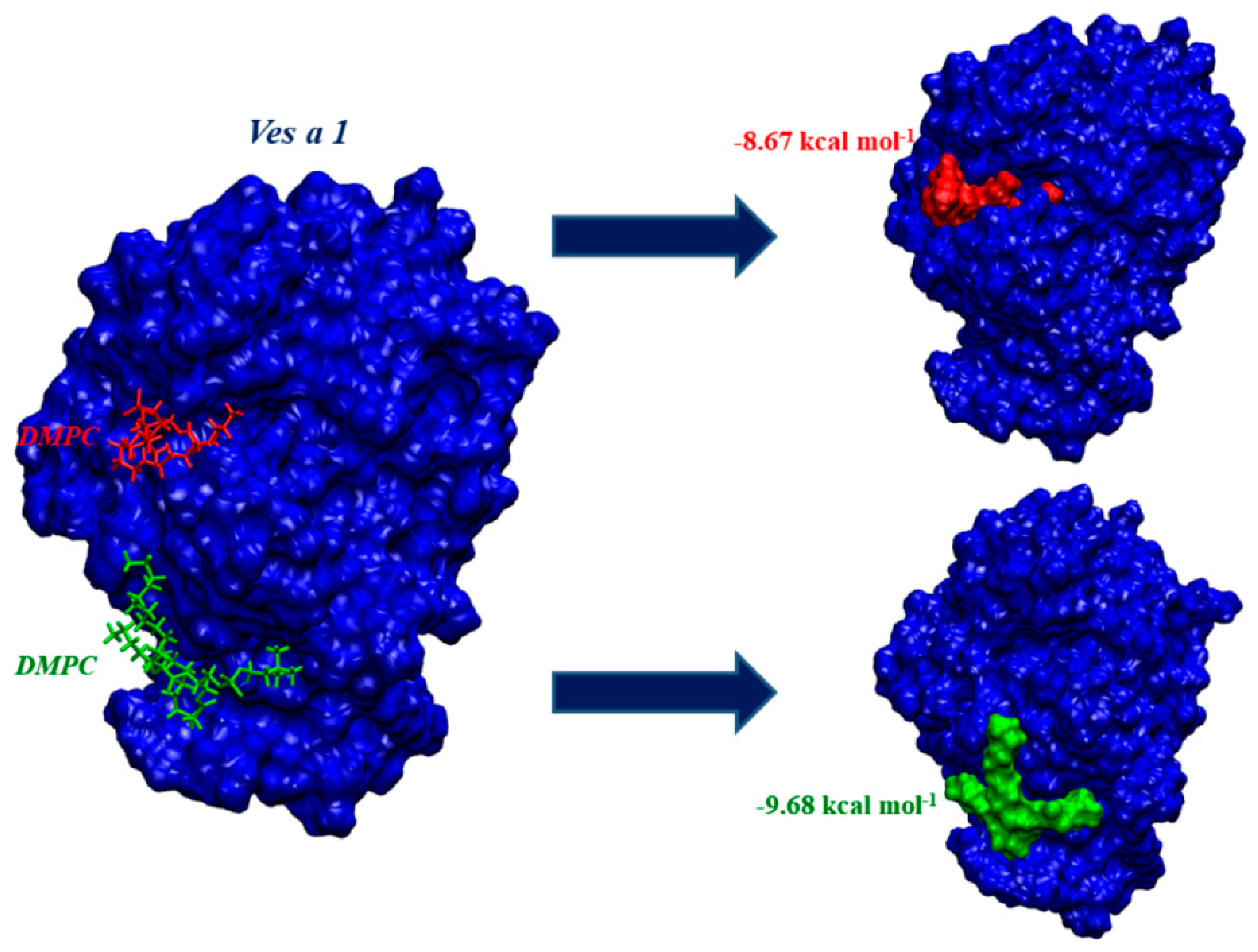
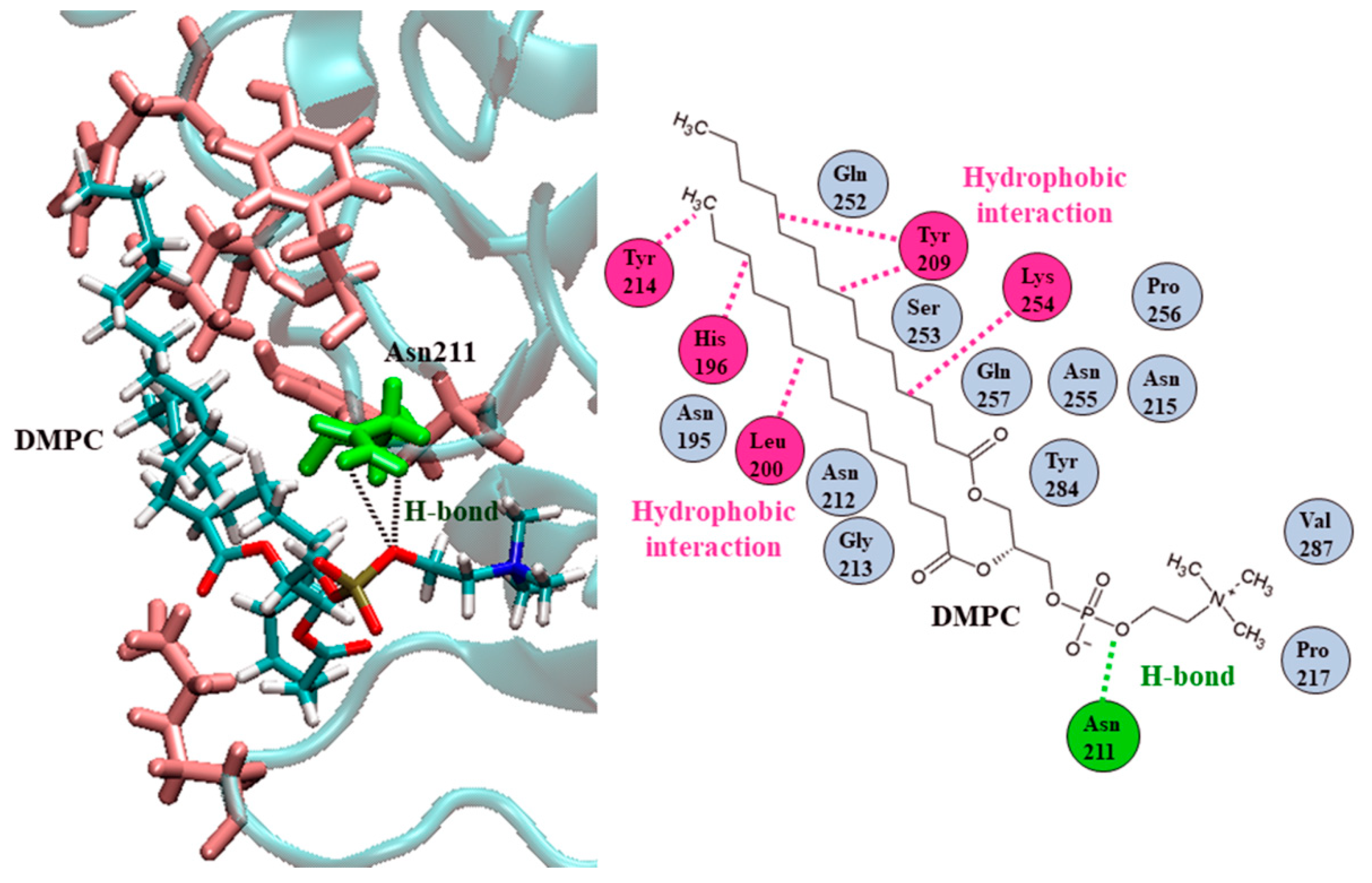
2.3. Ves a 1 Auxiliary Binding Site Identification and Interaction with Phospholipid
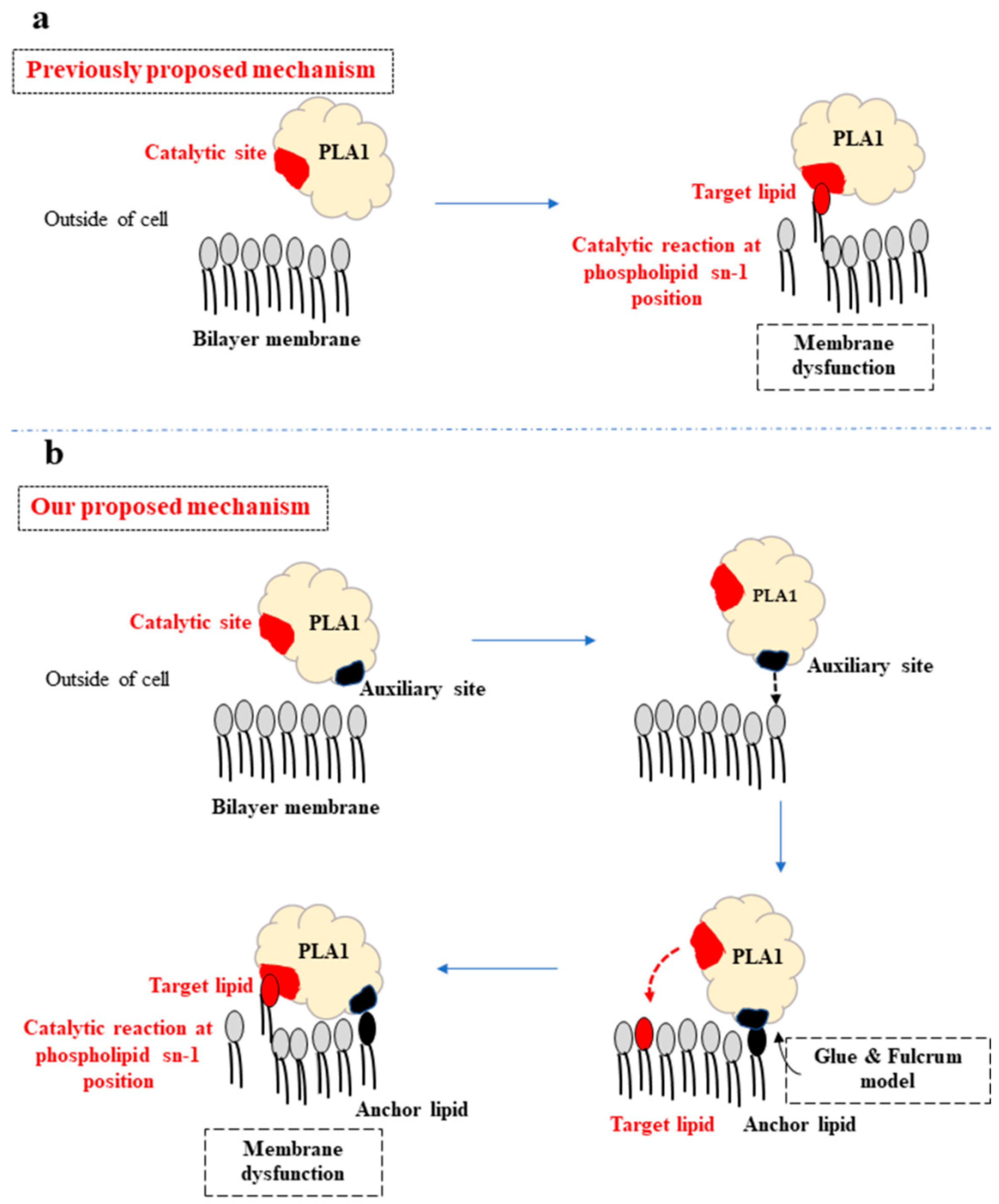
3. Discussion
4. Materials and Methods
4.1. Phospholipase A1 Structure Preparation
4.2. Molecular Dynamics Simulation of Ves a 1 and Molecular Docking of a DMPC-Ves a 1 Complex
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nguyen, L.T.; Saito, F.; Kojima, J.; Carpenter, J.M. Vespidae of Viet Nam (Insecta: Hymenoptera) 2. Taxonomic notes on Vespinae. Zoolog. Sci. 2006, 23, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Steen, C.J.; Janniger, C.K.; Schutzer, S.E.; Schwartz, R.A. Insect sting reactions to bees, wasps, and ants. Int. J. Dermatol. 2005, 44, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Das, R.N.; Mukherjee, K. Asian wasp envenomation and acute renal failure: A report of two cases. Mcgill. J. Med. 2008, 11, 25–28. [Google Scholar] [PubMed]
- Ullah, P.; Chowdhury, A.; Isha, I.T.; Mahmood, S.; Chowdhury, F.R.; Zeesan, U.A.M.; Manna, A.A.; Patwary, M.I. Wasp stings (Vespa affinis) induced acute kidney injury following rhabdomyolysis in a 25-year-old woman. J. Emerg. Pract. Trauma 2016, 2, 55–57. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, K.T.; Flood, A.A. Hymenoptera stings. Clin. Tech. Small Anim. Pract. 2006, 21, 194–204. [Google Scholar] [CrossRef] [PubMed]
- King, T.P.; Kochoumian, L.; Joslyn, A. Wasp venom proteins: Phospholipase A1 and B. Arch. Biochem. Biophys. 1984, 230, 1–12. [Google Scholar] [CrossRef]
- Yoon, K.A.; Kim, K.; Nguyen, P.; Seo, J.B.; Park, Y.H.; Kim, K.G.; Seo, H.Y.; Koh, Y.H.; Lee, S.H. Comparative functional venomics of social hornets Vespa crabro and Vespa analis. J. Asia-Pac. Entomol. 2015, 18, 815–823. [Google Scholar] [CrossRef]
- Sukprasert, S.; Rungsa, P.; Uawonggul, N.; Incamnoi, P.; Thammasirirak, S.; Daduang, J.; Daduang, S. Purification and structural characterisation of phospholipase A1 (Vespapase, Ves a 1) from Thai banded tiger wasp (Vespa affinis) venom. Toxicon 2013, 61, 151–164. [Google Scholar] [CrossRef]
- Aoki, J.; Inoue, A.; Makide, K.; Saiki, N.; Arai, H. Structure and function of extracellular phospholipase A1 belonging to the pancreatic lipase gene family. Biochimie 2007, 8, 197–204. [Google Scholar] [CrossRef]
- Hou, M.H.; Chuang, C.Y.; Ko, T.P.; Hu, N.J.; Chou, C.C.; Shih, Y.P.; Ho, C.L.; Wang, A.H.J. Crystal structure of vespid phospholipase A(1) reveals insights into the mechanism for cause of membrane dysfunction. Insect Biochem. Mol. Biol. 2016, 68, 79–88. [Google Scholar] [CrossRef]
- Perez-Riverol, A.; Lasa, A.M.; dos Santos-Pinto, J.R.A.; Palma, M.S. Insect venom phospholipase A1 and A2: Roles in the envenoming process and allergy. Insect Biochem. Mol. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Carrière, F.; Withers-Martinez, C.; van Tilbeurgh, H.; Roussel, A.; Cambillau, C.F.; Verger, R. Structural basis for the substrate selectivity of pancreatic lipases and some related proteins. Biochim. Biophys. Acta 1998, 1376, 417–432. [Google Scholar] [CrossRef]
- Lee, A.C.; Harris, J.L.; Kanna, K.K.; Hong, H.H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, A.C.; Magarkar, A.; Horta, M.; Lima, J.L.; Bunker, A.; Nunes, C.; Reis, S. Influence of doxorubicin on model cell membrane properties: Insights from in vitro and in silico studies. Sci. Rep. 2017, 7, 6343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nittayacharn, P.; Abenojar, E.; De Leon, A.; Wegierak, D.; Exner, A.A. Increasing Doxorubicin Loading in Lipid-Shelled Perfluoropropane Nanobubbles via a Simple Deprotonation Strategy. Front. Pharmacol. 2020, 11, 644. [Google Scholar] [CrossRef]
- Lu, H.; Marti, J. Cellular absorption of small molecules: Free energy landscapes of melatonin binding at phospholipid membranes. Sci. Rep. 2020, 10, 9235. [Google Scholar] [CrossRef]
- Stark, M.; Silva, T.F.D.; Levin, G.; Machuqueiro, M.; Assaraf, Y.G. The Lysosomotropic Activity of Hydrophobic Weak Base Drugs is Mediated via Their Intercalation into the Lysosomal Membrane. Cells 2020, 9, 1082. [Google Scholar] [CrossRef]
- Toroz, D.; Gould, I.R. A computational study of Anthracyclines interacting with lipid bilayers: Correlation of membrane insertion rates, orientation effects and localization with cytotoxicity. Sci. Rep. 2019, 9, 2155. [Google Scholar] [CrossRef]
- Marsh, D. Lipid-binding proteins: Structure of the phospholipid ligands. Protein Sci. 2003, 12, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Rungsa, P.; Incamnoi, P.; Sukprasert, S.; Uawonggul, N.; Klaynongsruang, S.; Daduang, J.; Patramanon, R.; Roytrakul, S.; Daduang, S. Comparative proteomic analysis of two wasps venom, Vespa tropica and Vespa affinis. Toxicon 2016, 119, 159–167. [Google Scholar] [CrossRef]
- Aggarwal, G.; Zarrow, J.E.; Mashhadi, Z.; Flynn, C.R.; Vinson, P.; Weaver, C.D.; Davies, S.S. Symmetrically substituted dichlorophenes inhibit N-acyl-phosphatidylethanolamine phospholipase D. J. Bio. Chem. 2020, 295, 7289–7300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Alaoui, M.; Soulère, L.; Noiriel, A.; Popowycz, F.; Khatib, A.; Queneau, Y.; Abousalham, A. A continuous spectrophotometric assay that distinguishes between phospholipase A1 and A2 activities. J. Bio. Chem. 2016, 57, 1589–1597. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchan, D.W.A.; Minneci, F.; Nugent, T.C.O.; Bryson, K.; Jones, D.T. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013, 41, W349–W357. [Google Scholar] [CrossRef]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Petersen, B.; Petersen, T.N.; Andersen, P.; Nielsen, M.; Lundegaard, C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 2009, 9, 51. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Leman, J.K.; Weitzner, B.D.; Lewis, S.M.; Adolf-Bryfogle, J.; Alam, N.; Alford, R.F.; Aprahamian, M.; Baker, D.; Barlow, K.A.; Barth, P.; et al. Macromolecular modeling and design in Rosetta: Recent methods and frameworks. Nat. Methods 2020. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK - a program to check the stereochemical quality of protein structures. J. App. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Song, L.F.; Lee, T.-S.; Zhu, C.; York, D.M.; Merz, K.M. Using AMBER18 for Relative Free Energy Calculations. J. Chem. Inf. Model. 2019, 59, 3128–3135. [Google Scholar] [CrossRef] [PubMed]
- Pastor, R.W.; Brooks, B.R.; Szabo, A. An analysis of the accuracy of Langevin and molecular dynamics algorithms. Mol. Phys. 1988, 65, 1409–1419. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. AMBER 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [Green Version]
- BIOVIA, Dassault Systèmes Discovery Studio Visualizer, Release 2019, San Diego, CA, USA, 2019. Available online: https://discover.3ds.com/discovery-studio-visualizer-download (accessed on 20 January 2020).
- ACD/ChemSketch, version 2019.2.2; Advanced Chemistry Development, Inc.: Toronto, ON, Canada; Available online: www.acdlabs.com (accessed on 30 December 2019).










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teajaroen, W.; Phimwapi, S.; Daduang, J.; Klaynongsruang, S.; Tipmanee, V.; Daduang, S. A Role of Newly Found Auxiliary Site in Phospholipase A1 from Thai Banded Tiger Wasp (Vespa affinis) in Its Enzymatic Enhancement: In Silico Homology Modeling and Molecular Dynamics Insights. Toxins 2020, 12, 510. https://doi.org/10.3390/toxins12080510
Teajaroen W, Phimwapi S, Daduang J, Klaynongsruang S, Tipmanee V, Daduang S. A Role of Newly Found Auxiliary Site in Phospholipase A1 from Thai Banded Tiger Wasp (Vespa affinis) in Its Enzymatic Enhancement: In Silico Homology Modeling and Molecular Dynamics Insights. Toxins. 2020; 12(8):510. https://doi.org/10.3390/toxins12080510
Chicago/Turabian StyleTeajaroen, Withan, Suphaporn Phimwapi, Jureerut Daduang, Sompong Klaynongsruang, Varomyalin Tipmanee, and Sakda Daduang. 2020. "A Role of Newly Found Auxiliary Site in Phospholipase A1 from Thai Banded Tiger Wasp (Vespa affinis) in Its Enzymatic Enhancement: In Silico Homology Modeling and Molecular Dynamics Insights" Toxins 12, no. 8: 510. https://doi.org/10.3390/toxins12080510
APA StyleTeajaroen, W., Phimwapi, S., Daduang, J., Klaynongsruang, S., Tipmanee, V., & Daduang, S. (2020). A Role of Newly Found Auxiliary Site in Phospholipase A1 from Thai Banded Tiger Wasp (Vespa affinis) in Its Enzymatic Enhancement: In Silico Homology Modeling and Molecular Dynamics Insights. Toxins, 12(8), 510. https://doi.org/10.3390/toxins12080510