Undercover Agents of Infection: The Stealth Strategies of T4SS-Equipped Bacterial Pathogens


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
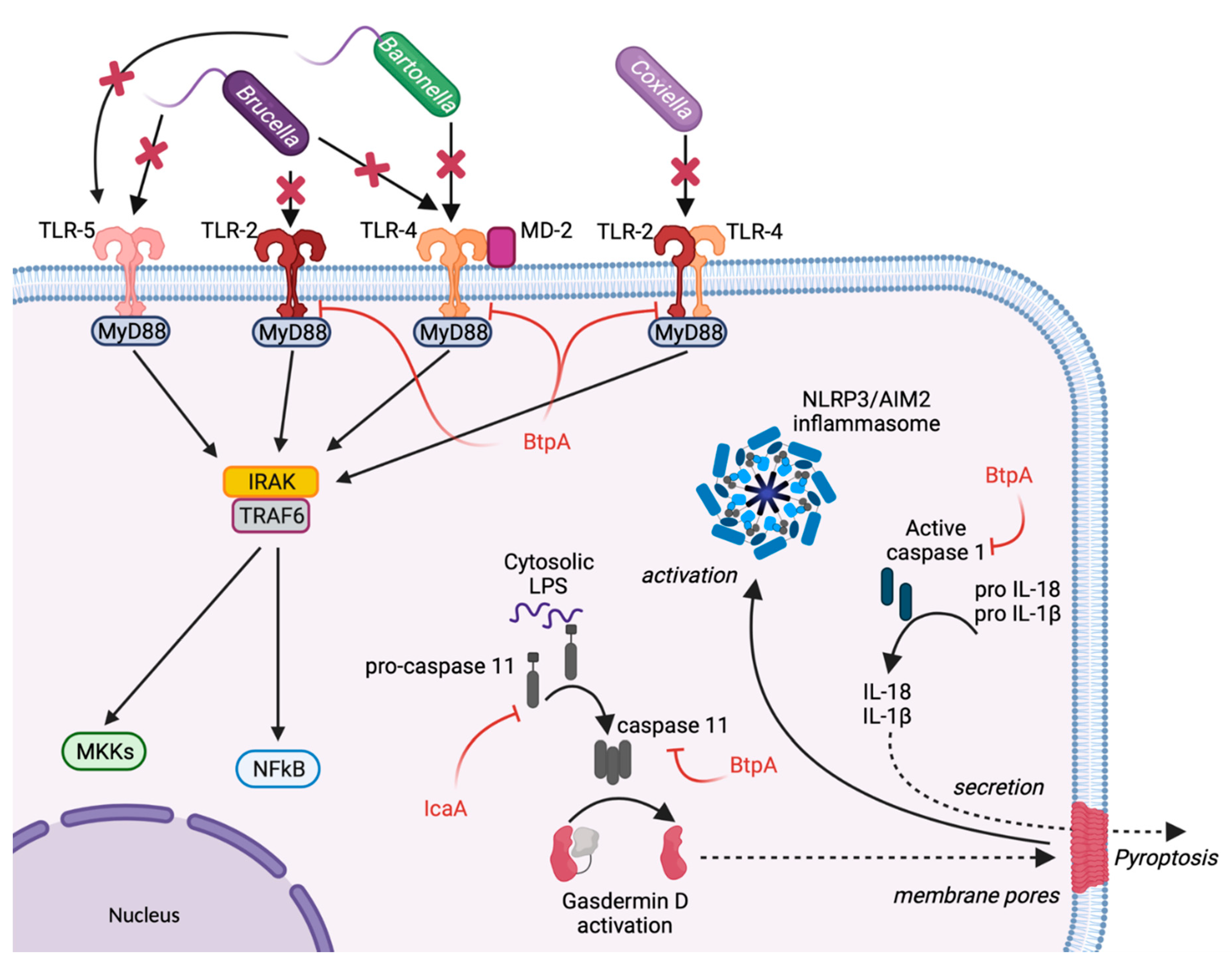
2. Escape from Host Sensing
3. Inhibition of Inflammasome
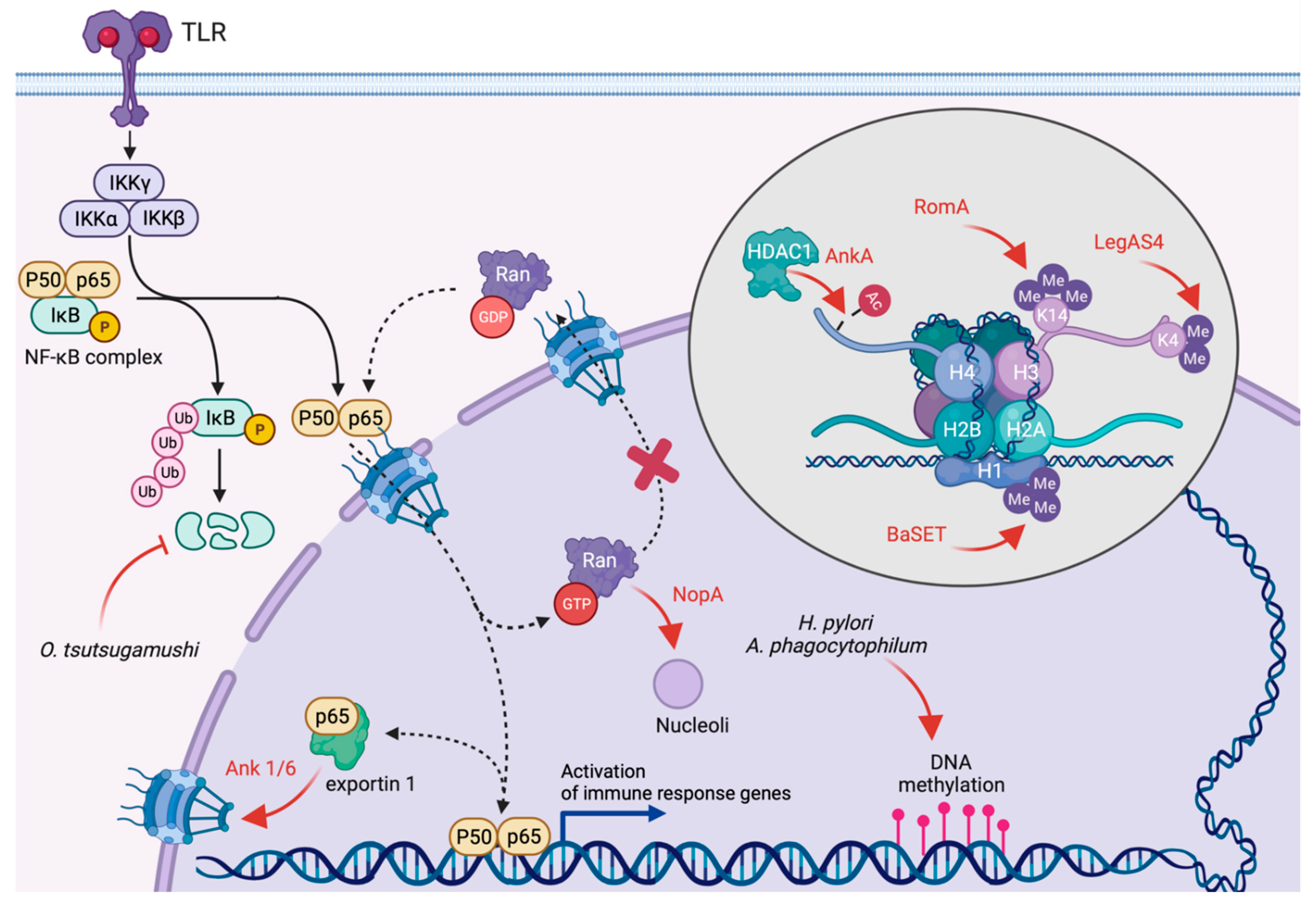
4. Inhibition of Transcription and Translation
5. Modulation of the Unfolded Protein Response
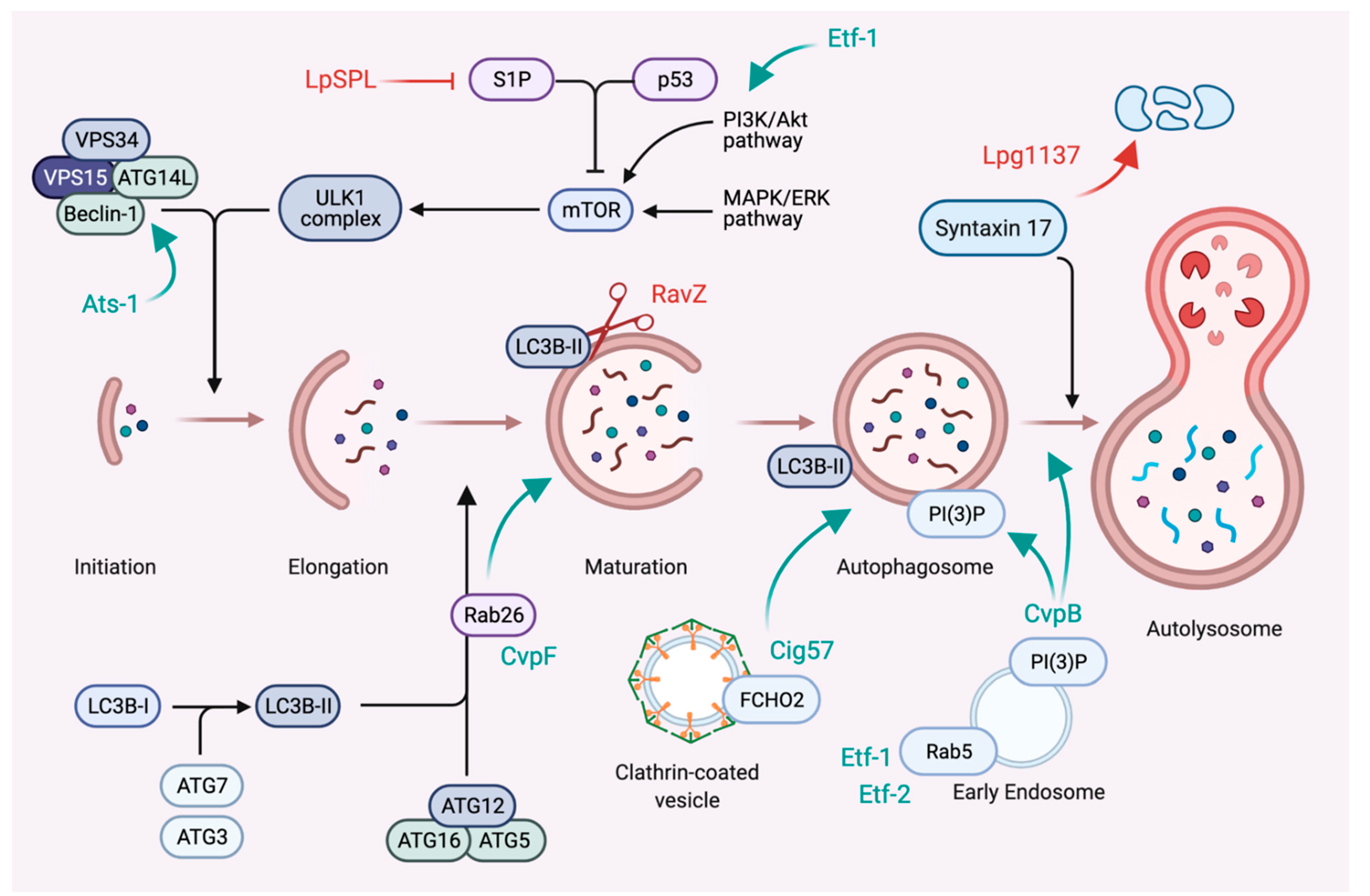
6. Subversion of Autophagy
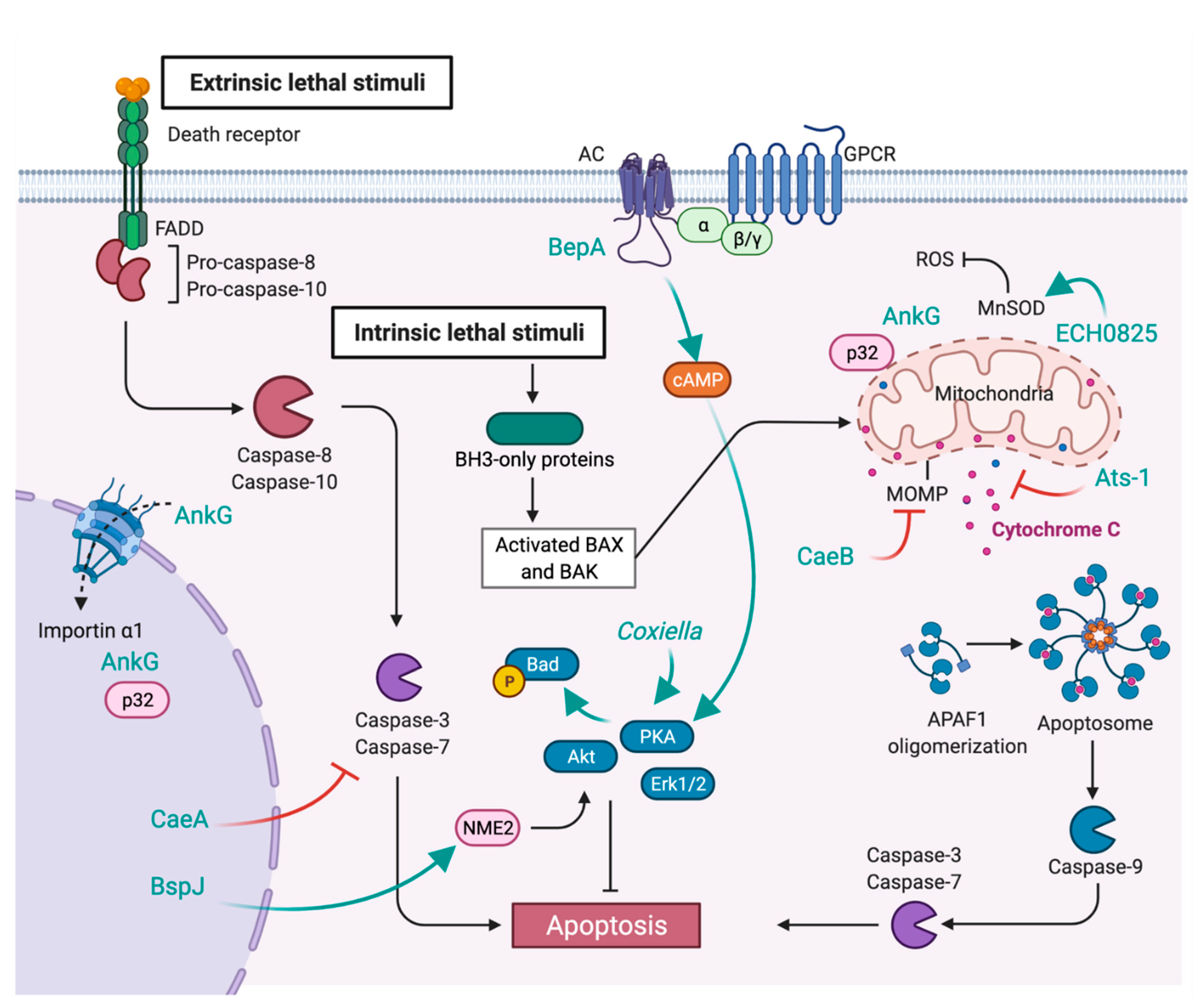
7. Inhibition of Apoptosis
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Green, E.R.; Mecsas, J. Bacterial Secretion Systems: An Overview. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallden, K.; Rivera-Calzada, A.; Waksman, G. Microreview: Type IV Secretion Systems: Versatility and Diversity in Function. Cell. Microbiol. 2010, 12, 1203–1212. [Google Scholar] [CrossRef] [Green Version]
- Costa, T.R.D.; Felisberto-Rodrigues, C.; Meir, A.; Prevost, M.S.; Redzej, A.; Trokter, M.; Waksman, G. Secretion Systems in Gram-Negative Bacteria: Structural and Mechanistic Insights. Nat. Rev. Microbiol. 2015, 13, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.; Tegtmeyer, N.; Stingl, K.; Backert, S. Four Chromosomal Type IV Secretion Systems in Helicobacter pylori: Composition, Structure and Function. Front. Microbiol. 2020, 11, 1592. [Google Scholar] [CrossRef]
- Li, Y.G.; Christie, P.J. Agrobacterium Biology, from Basic Science to Biotechnology. Curr. Top Microbiol. 2018, 418, 233–260. [Google Scholar] [CrossRef]
- Gonzalez-Rivera, C.; Bhatty, M.; Christie, P.J. Mechanism and Function of Type IV Secretion during Infection of the Human Host. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, A.; Dehio, C. Role of Distinct Type-IV-secretion Systems and Secreted Effector Sets in Host Adaptation by Pathogenic Bartonella Species. Cell. Microbiol. 2019, 21, e13004. [Google Scholar] [CrossRef] [Green Version]
- Kubori, T.; Nagai, H. The Type IVB Secretion System: An Enigmatic Chimera. Curr. Opin. Microbiol. 2016, 29, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Martinez, E.; Siadous, F.A.; Bonazzi, M. Tiny Architects: Biogenesis of Intracellular Replicative Niches by Bacterial Pathogens. FEMS Microbiol. Rev. 2018, 42, 425–447. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.C.; Broz, P. Sensing of Invading Pathogens by GBPs: At the Crossroads between Cell-autonomous and Innate Immunity. J. Leukoc. Biol. 2018, 104, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Coers, J. Self and Non-Self Discrimination of Intracellular Membranes by the Innate Immune System. PLoS Pathog. 2013, 9, e1003538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Ingle, H.; Prasad, D.V.R.; Kumar, H. Recognition of Bacterial Infection by Innate Immune Sensors. Crit. Rev. Microbiol. 2012, 39, 229–246. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [Green Version]
- Ben-Tekaya, H.; Gorvel, J.-P.; Dehio, C. Bartonella and Brucella—Weapons and Strategies for Stealth Attack. Csh. Perspect. Med. 2013, 3, a010231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.I.; Ernst, R.K.; Bader, M.W. LPS, TLR4 and Infectious Disease Diversity. Nat. Rev. Microbiol. 2005, 3, 36–46. [Google Scholar] [CrossRef]
- Popa, C.; Abdollahi-Roodsaz, S.; Joosten, L.A.B.; Takahashi, N.; Sprong, T.; Matera, G.; Liberto, M.C.; Foca, A.; van Deuren, M.; Kullberg, B.J.; et al. Bartonella quintana Lipopolysaccharide Is a Natural Antagonist of Toll-Like Receptor 4. Infect. Immun. 2007, 75, 4831–4837. [Google Scholar] [CrossRef] [Green Version]
- Malgorzata-Miller, G.; Heinbockel, L.; Brandenburg, K.; van der Meer, J.W.M.; Netea, M.G.; Joosten, L.A.B. Bartonella quintana Lipopolysaccharide (LPS): Structure and Characteristics of a Potent TLR4 Antagonist for in-Vitro and in-Vivo Applications. Sci. Rep. 2016, 6, 34221. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.A. Brucella Lipopolysaccharide and Pathogenicity: The Core of the Matter. Virulence 2018, 9, 379–382. [Google Scholar] [CrossRef] [Green Version]
- Conde-Álvarez, R.; Arce-Gorvel, V.; Iriarte, M.; Manček-Keber, M.; Barquero-Calvo, E.; Palacios-Chaves, L.; Chacón-Díaz, C.; Chaves-Olarte, E.; Martirosyan, A.; von Bargen, K.; et al. The Lipopolysaccharide Core of Brucella abortus Acts as a Shield Against Innate Immunity Recognition. PLoS Pathog. 2012, 8, e1002675. [Google Scholar] [CrossRef] [Green Version]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.-S.; Lee, H.; Lee, J.-O. The Structural Basis of Lipopolysaccharide Recognition by the TLR4–MD-2 Complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Conti, F.; Boucherit, N.; Baldassarre, V.; Trouplin, V.; Toman, R.; Mottola, G.; Mege, J.-L.; Ghigo, E. Coxiella burnetii Lipopolysaccharide Blocks P38α-MAPK Activation through the Disruption of TLR-2 and TLR-4 Association. Front. Cell. Infect. Microbiol. 2015, 4, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terwagne, M.; Ferooz, J.; Rolán, H.G.; Sun, Y.; Atluri, V.; Xavier, M.N.; Franchi, L.; Núñez, G.; Legrand, T.; Flavell, R.A.; et al. Innate Immune Recognition of Flagellin Limits Systemic Persistence of Brucella. Cell. Microbiol. 2013, 15, 942–960. [Google Scholar] [CrossRef] [Green Version]
- Andersen-Nissen, E.; Smith, K.D.; Strobe, K.L.; Barrett, S.L.R.; Cookson, B.T.; Logan, S.M.; Aderem, A. Evasion of Toll-like Receptor 5 by Flagellated Bacteria. Proc. Natl. Acad. Sci. USA 2005, 102, 9247–9252. [Google Scholar] [CrossRef] [Green Version]
- Snyder, G.A.; Deredge, D.; Waldhuber, A.; Fresquez, T.; Wilkins, D.Z.; Smith, P.T.; Durr, S.; Cirl, C.; Jiang, J.; Jennings, W.; et al. Crystal Structures of the Toll/Interleukin-1 Receptor (TIR) Domains from the Brucella Protein TcpB and Host Adaptor TIRAP Reveal Mechanisms of Molecular Mimicry. J. Biol. Chem. 2014, 289, 669–679. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, D.; Koblansky, A.; Gaines, J.; Brown, T.; West, A.P.; Zhang, D.; Nishikawa, T.; Park, S.-G.; Roop, R.M.; Ghosh, S. Subversion of Innate Immune Responses by Brucella through the Targeted Degradation of the TLR Signaling Adapter, MAL. J. Immunol. 2010, 184, 956–964. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, G.K.; Yu, Q.; Harms, J.S.; Splitter, G.A. Brucella TIR Domain-Containing Protein Mimics Properties of the Toll-like Receptor Adaptor Protein TIRAP. J. Biol. Chem. 2009, 284, 9892–9898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salcedo, S.P.; Marchesini, M.I.; Degos, C.; Terwagne, M.; Bargen, K.V.; Lepidi, H.; Herrmann, C.K.; Lacerda, T.L.S.; Imbert, P.R.C.; Pierre, P.; et al. BtpB, a Novel Brucella TIR-Containing Effector Protein with Immune Modulatory Functions. Front. Cell. Infect. Microbiol. 2013, 3, 28. [Google Scholar] [CrossRef] [Green Version]
- Coronas-Serna, J.M.; Louche, A.; Rodríguez-Escudero, M.; Roussin, M.; Imbert, P.R.C.; Rodríguez-Escudero, I.; Terradot, L.; Molina, M.; Gorvel, J.-P.; Cid, V.J.; et al. The TIR-Domain Containing Effectors BtpA and BtpB from Brucella abortus Impact NAD Metabolism. PLoS Pathog. 2020, 16, e1007979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krakauer, T. Inflammasomes, Autophagy, and Cell Death: The Trinity of Innate Host Defense against Intracellular Bacteria. Mediat. Inflamm. 2019, 2019, 2471215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Shao, F. SnapShot: The Noncanonical Inflammasome. Cell 2017, 168, 544. [Google Scholar] [CrossRef]
- Broz, P. Recognition of Intracellular Bacteria by Inflammasomes. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Jakka, P.; Namani, S.; Murugan, S.; Rai, N.; Radhakrishnan, G. The Brucella Effector Protein TcpB Induces Degradation of Inflammatory Caspases and Thereby Subverts Non-Canonical Inflammasome Activation in Macrophages. J. Biol. Chem. 2017, 292, 20613–20627. [Google Scholar] [CrossRef] [Green Version]
- Cunha, L.D.; Ribeiro, J.M.; Fernandes, T.D.; Massis, L.M.; Khoo, C.A.; Moffatt, J.H.; Newton, H.J.; Roy, C.R.; Zamboni, D.S. Inhibition of Inflammasome Activation by Coxiella burnetii Type IV Secretion System Effector IcaA. Nat. Commun. 2015, 6, 10205. [Google Scholar] [CrossRef]
- Martinez, E.; Cantet, F.; Fava, L.; Norville, I.; Bonazzi, M. Identification of OmpA, a Coxiella burnetii Protein Involved in Host Cell Invasion, by Multi-Phenotypic High-Content Screening. PLoS Pathog. 2014, 10, e1004013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platanitis, E.; Decker, T. Regulatory Networks Involving STATs, IRFs, and NFκB in Inflammation. Front. Immunol. 2018, 9, 2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Wang, G.; Qian, J. Transcription Factors as Readers and Effectors of DNA Methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Maekita, T.; Nakazawa, K.; Mihara, M.; Nakajima, T.; Yanaoka, K.; Iguchi, M.; Arii, K.; Kaneda, A.; Tsukamoto, T.; Tatematsu, M.; et al. High Levels of Aberrant DNA Methylation in Helicobacter pylori–Infected Gastric Mucosae and Its Possible Association with Gastric Cancer Risk. Clin. Cancer Res. 2006, 12, 989–995. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, S.H.G.; Yegnasubramanian, S.; Dumler, J.S. Global DNA Methylation Changes and Differential Gene Expression in Anaplasma phagocytophilum-Infected Human Neutrophils. Clin. Epigenet. 2015, 7, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabiec, A.M.; Potempa, J. Epigenetic Regulation in Bacterial Infections: Targeting Histone Deacetylases. Crit. Rev. Microbiol. 2017, 44, 336–350. [Google Scholar] [CrossRef] [Green Version]
- Rolando, M.; Sanulli, S.; Rusniok, C.; Gomez-Valero, L.; Bertholet, C.; Sahr, T.; Margueron, R.; Buchrieser, C. Legionella pneumophila Effector RomA Uniquely Modifies Host Chromatin to Repress Gene Expression and Promote Intracellular Bacterial Replication. Cell Host Microbe 2013, 13, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Lu, Q.; Wang, G.; Xu, H.; Huang, H.; Cai, T.; Kan, B.; Ge, J.; Shao, F. SET-domain Bacterial Effectors Target Heterochromatin Protein 1 to Activate Host RDNA Transcription. EMBO Rep. 2013, 14, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Cazalet, C.; Rusniok, C.; Brüggemann, H.; Zidane, N.; Magnier, A.; Ma, L.; Tichit, M.; Jarraud, S.; Bouchier, C.; Vandenesch, F.; et al. Evidence in the Legionella pneumophila Genome for Exploitation of Host Cell Functions and High Genome Plasticity. Nat. Genet. 2004, 36, 1165–1173. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.D.; Swanson, M.S. Comparative Analysis of Legionella pneumophila and Legionella micdadei Virulence Traits. Infect. Immun. 1999, 67, 4134–4142. [Google Scholar] [CrossRef] [Green Version]
- Mujtaba, S.; Winer, B.Y.; Jaganathan, A.; Patel, J.; Sgobba, M.; Schuch, R.; Gupta, Y.K.; Haider, S.; Wang, R.; Fischetti, V.A. Anthrax SET Protein a potential virulence determinant that epigenetically represses NF-kB activation in infected macrophages. J. Biol. Chem. 2013, 288, 23458–23472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennini, M.E.; Perrinet, S.; Dautry-Varsat, A.; Subtil, A. Histone Methylation by NUE, a Novel Nuclear Effector of the Intracellular Pathogen Chlamydia trachomatis. PLoS Pathog. 2010, 6, e1000995. [Google Scholar] [CrossRef]
- Rennoll-Bankert, K.E.; Garcia-Garcia, J.C.; Sinclair, S.H.; Dumler, J.S. Chromatin-bound Bacterial Effector Ankyrin A Recruits Histone Deacetylase 1 and Modifies Host Gene Expression. Cell. Microbiol. 2015, 17, 1640–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Garcia, J.C.; Rennoll-Bankert, K.E.; Pelly, S.; Milstone, A.M.; Dumler, J.S. Silencing of Host Cell CYBB Gene Expression by the Nuclear Effector AnkA of the Intracellular Pathogen Anaplasma phagocytophilum. Infect. Immun. 2009, 77, 2385–2391. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Garcia, J.C.; Barat, N.C.; Trembley, S.J.; Dumler, J.S. Epigenetic Silencing of Host Cell Defense Genes Enhances Intracellular Survival of the Rickettsial Pathogen Anaplasma phagocytophilum. PLoS Pathog. 2009, 5, e1000488. [Google Scholar] [CrossRef] [PubMed]
- Dorrington, M.G.; Fraser, I.D.C. NF-ΚB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef]
- Evans, S.M.; Rodino, K.G.; Adcox, H.E.; Carlyon, J.A. Orientia tsutsugamushi Uses Two Ank Effectors to Modulate NF-ΚB P65 Nuclear Transport and Inhibit NF-ΚB Transcriptional Activation. PLoS Pathog. 2018, 14, e1007023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wangsanut, T.; Brann, K.R.; Adcox, H.E.; Carlyon, J.A. Orientia tsutsugamushi Modulates Cellular Levels of NF-kB Inhibitor P105. PLoS Neglect. Trop. Dis. 2021, 15, e0009339. [Google Scholar] [CrossRef]
- Burette, M.; Allombert, J.; Lambou, K.; Maarifi, G.; Nisole, S.; Case, E.D.R.; Blanchet, F.P.; Hassen-Khodja, C.; Cabantous, S.; Samuel, J.; et al. Modulation of Innate Immune Signaling by a Coxiella burnetii Eukaryotic-like Effector Protein. Proc. Natl. Acad. Sci. USA 2020, 117, 13708–13718. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M. Molecular Mechanism of the Nuclear Protein Import Cycle. Nat. Rev. Mol. Cell Biol. 2007, 8, 195–208. [Google Scholar] [CrossRef]
- Hillmer, E.J.; Zhang, H.; Li, H.S.; Watowich, S.S. STAT3 Signaling in Immunity. Cytokine Growth Factor Rev. 2016, 31, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorg, I.; Schmutz, C.; Lu, Y.-Y.; Fromm, K.; Siewert, L.K.; Bögli, A.; Strack, K.; Harms, A.; Dehio, C. A Bartonella Effector Acts as Signaling Hub for Intrinsic STAT3 Activation to Trigger Anti-Inflammatory Responses. Cell Host Microbe 2020, 27, 476–485.e7. [Google Scholar] [CrossRef]
- Hetz, C. The Unfolded Protein Response: Controlling Cell Fate Decisions under ER Stress and Beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, Regulation and Functions of the Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- De Jong, M.F.; Starr, T.; Winter, M.G.; den Hartigh, A.B.; Child, R.; Knodler, L.A.; van Dijl, J.M.; Celli, J.; Tsolis, R.M. Sensing of Bacterial Type IV Secretion via the Unfolded Protein Response. Mbio 2013, 4, e00418-12. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.A.; Khan, M.; Magnani, D.D.; Harms, J.S.; Durward, M.; Radhakrishnan, G.K.; Liu, Y.-P.; Splitter, G.A. Brucella Induces an Unfolded Protein Response via TcpB That Supports Intracellular Replication in Macrophages. PLoS Pathog. 2013, 9, e1003785. [Google Scholar] [CrossRef] [Green Version]
- Hempstead, A.D.; Isberg, R.R. Inhibition of Host Cell Translation Elongation by Legionella pneumophila Blocks the Host Cell Unfolded Protein Response. Proc. Natl. Acad. Sci. USA 2015, 112, E6790–E6797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treacy-Abarca, S.; Mukherjee, S. Legionella Suppresses the Host Unfolded Protein Response via Multiple Mechanisms. Nat. Commun. 2015, 6, 7887. [Google Scholar] [CrossRef] [Green Version]
- Brann, K.R.; Fullerton, M.S.; Voth, D.E. Coxiella burnetii Requires Host Eukaryotic Initiation Factor 2α Activity for Efficient Intracellular Replication. Infect. Immun. 2020, 88, e00096-20. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, A.; Beare, P.A.; Schulze-Luehrmann, J.; Cordsmeier, A.; Pazen, T.; Sonnewald, S.; Lührmann, A. The Coxiella burnetii Effector Protein CaeB Modulates Endoplasmatic Reticulum (ER) Stress Signalling and Is Required for Efficient Replication in Galleria mellonella. Cell. Microbiol. 2021, 23, e13305. [Google Scholar] [CrossRef] [PubMed]
- Klingenbeck, L.; Eckart, R.A.; Berens, C.; Lührmann, A. The Coxiella burnetii Type IV Secretion System Substrate CaeB Inhibits Intrinsic Apoptosis at the Mitochondrial Level. Cell. Microbiol. 2013, 15, 675–687. [Google Scholar] [CrossRef]
- Lee, J.; Ye, Y. Bag6/Bat3/Scythe: A Novel Chaperone Activity with Diverse Regulatory Functions in Protein Biogenesis and Degradation. Bioessays 2013, 35, 377–385. [Google Scholar] [CrossRef]
- Rodino, K.G.; VieBrock, L.; Evans, S.M.; Ge, H.; Richards, A.L.; Carlyon, J.A. Orientia tsutsugamushi Modulates Endoplasmic Reticulum-Associated Degradation to Benefit Its Growth. Infect. Immun. 2018, 86, e00596-17. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and Regulation. Microb. Cell 2016, 3, 588. [Google Scholar] [CrossRef]
- Choy, A.; Dancourt, J.; Mugo, B.; O’Connor, T.J.; Isberg, R.R.; Melia, T.J.; Roy, C.R. The Legionella Effector RavZ Inhibits Host Autophagy Through Irreversible Atg8 Deconjugation. Science 2012, 338, 1072–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arasaki, K.; Mikami, Y.; Shames, S.R.; Inoue, H.; Wakana, Y.; Tagaya, M. Legionella Effector Lpg1137 Shuts down ER-Mitochondria Communication through Cleavage of Syntaxin 17. Nat. Commun. 2017, 8, 15406. [Google Scholar] [CrossRef] [PubMed]
- Rolando, M.; Escoll, P.; Nora, T.; Botti, J.; Boitez, V.; Bedia, C.; Daniels, C.; Abraham, G.; Stogios, P.J.; Skarina, T.; et al. Legionella pneumophila S1P-Lyase Targets Host Sphingolipid Metabolism and Restrains Autophagy. Proc. Natl. Acad. Sci. USA 2016, 113, 1901–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, H.; Xiong, Q.; Yamamoto, A.; Hayashi-Nishino, M.; Rikihisa, Y. Autophagosomes Induced by a Bacterial Beclin 1 Binding Protein Facilitate Obligatory Intracellular Infection. Proc. Natl. Acad. Sci. USA 2012, 109, 20800–20807. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Liu, H.; Xiong, Q.; Niu, H.; Cheng, Z.; Yamamoto, A.; Rikihisa, Y. Ehrlichia Secretes Etf-1 to Induce Autophagy and Capture Nutrients for Its Growth through RAB5 and Class III Phosphatidylinositol 3-Kinase. Autophagy 2016, 12, 2145–2166. [Google Scholar] [CrossRef] [Green Version]
- Yan, Q.; Lin, M.; Huang, W.; Teymournejad, O.; Johnson, J.M.; Hays, F.A.; Liang, Z.; Li, G.; Rikihisa, Y. Ehrlichia Type IV Secretion System Effector Etf-2 Binds to Active RAB5 and Delays Endosome Maturation. Proc. Natl. Acad. Sci. USA 2018, 115, 201806904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, M.G.; Vázquez, C.L.; Munafó, D.B.; Zoppino, F.C.M.; Berón, W.; Rabinovitch, M.; Colombo, M.I. Autophagy Induction Favours the Generation and Maturation of the Coxiella-replicative Vacuoles. Cell. Microbiol. 2005, 7, 981–993. [Google Scholar] [CrossRef]
- Newton, P.; Thomas, D.R.; Reed, S.C.O.; Lau, N.; Xu, B.; Ong, S.Y.; Pasricha, S.; Madhamshettiwar, P.B.; Edgington-Mitchell, L.E.; Simpson, K.J.; et al. Lysosomal Degradation Products Induce Coxiella burnetii Virulence. Proc. Natl. Acad. Sci. USA 2020, 117, 6801–6810. [Google Scholar] [CrossRef] [Green Version]
- Newton, H.J.; Kohler, L.J.; McDonough, J.A.; Temoche-Diaz, M.; Crabill, E.; Hartland, E.L.; Roy, C.R. A Screen of Coxiella burnetii Mutants Reveals Important Roles for Dot/Icm Effectors and Host Autophagy in Vacuole Biogenesis. PLoS Pathog. 2014, 10, e1004286. [Google Scholar] [CrossRef] [Green Version]
- Kohler, L.J.; Reed, S.R.; Sarraf, S.A.; Arteaga, D.D.; Newton, H.J.; Roy, C.R. Effector Protein Cig2 Decreases Host Tolerance of Infection by Directing Constitutive Fusion of Autophagosomes with the Coxiella-Containing Vacuole. Mbio 2016, 7, e01127-16. [Google Scholar] [CrossRef] [Green Version]
- Martinez, E.; Allombert, J.; Cantet, F.; Lakhani, A.; Yandrapalli, N.; Neyret, A.; Norville, I.H.; Favard, C.; Muriaux, D.; Bonazzi, M. Coxiella burnetii Effector CvpB Modulates Phosphoinositide Metabolism for Optimal Vacuole Development. Proc. Natl. Acad. Sci. USA 2016, 113, E3260–E3269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siadous, F.A.; Cantet, F.; Schaik, E.V.; Burette, M.; Allombert, J.; Lakhani, A.; Bonaventure, B.; Goujon, C.; Samuel, J.; Bonazzi, M.; et al. Coxiella Effector Protein CvpF Subverts RAB26-Dependent Autophagy to Promote Vacuole Biogenesis and Virulence. Autophagy 2020, 17, 706–722. [Google Scholar] [CrossRef] [Green Version]
- Latomanski, E.A.; Newton, P.; Khoo, C.A.; Newton, H.J. The Effector Cig57 Hijacks FCHO-Mediated Vesicular Trafficking to Facilitate Intracellular Replication of Coxiella burnetii. PLoS Pathog. 2016, 12, e1006101. [Google Scholar] [CrossRef]
- Latomanski, E.A.; Newton, H.J. Interaction between Autophagic Vesicles and the Coxiella-Containing Vacuole Requires CLTC (Clathrin Heavy Chain). Autophagy 2018, 14, 1710–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starr, T.; Child, R.; Wehrly, T.D.; Hansen, B.; Hwang, S.; López-Otin, C.; Virgin, H.W.; Celli, J. Selective Subversion of Autophagy Complexes Facilitates Completion of the Brucella Intracellular Cycle. Cell Host Microbe 2012, 11, 33–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, Y.; Li, H.; Song, H.; Zhai, N.; Lou, L.; Wang, F.; Zhang, K.; Bao, W.; Jin, X.; et al. Brucella Dysregulates Monocytes and Inhibits Macrophage Polarization through LC3-Dependent Autophagy. Front. Immunol. 2017, 8, 691. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, Pyroptosis and Apoptosis: An Intricate Game of Cell Death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Voth, D.E.; Howe, D.; Heinzen, R.A. Coxiella burnetii Inhibits Apoptosis in Human THP-1 Cells and Monkey Primary Alveolar Macrophages. Infect. Immun. 2007, 75, 4263–4271. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, L.J.; Graham, J.G.; Kurten, R.C.; Voth, D.E. Coxiella burnetii Exploits Host CAMP-dependent Protein Kinase Signalling to Promote Macrophage Survival. Cell. Microbiol. 2014, 16, 146–159. [Google Scholar] [CrossRef] [Green Version]
- Voth, D.E.; Heinzen, R.A. Sustained Activation of Akt and Erk1/2 Is Required for Coxiella burnetii Antiapoptotic Activity. Infect. Immun. 2009, 77, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Lührmann, A.; Nogueira, C.V.; Carey, K.L.; Roy, C.R. Inhibition of Pathogen-Induced Apoptosis by a Coxiella burnetii Type IV Effector Protein. Proc. Natl. Acad. Sci. USA 2010, 107, 18997–19001. [Google Scholar] [CrossRef] [Green Version]
- Eckart, R.A.; Bisle, S.; Schulze-Luehrmann, J.; Wittmann, I.; Jantsch, J.; Schmid, B.; Berens, C.; Lührmann, A. Antiapoptotic Activity of Coxiella burnetii Effector Protein AnkG Is Controlled by P32-Dependent Trafficking. Infect. Immun. 2014, 82, 2763–2771. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, W.; Eckart, R.A.; Schmid, B.; Cagköylü, H.; Hof, K.; Muller, Y.A.; Amin, B.; Lührmann, A. Nuclear Trafficking of the Anti-apoptotic Coxiella burnetii Effector Protein AnkG Requires Binding to P32 and Importin-α1. Cell. Microbiol. 2016, 19, e12634. [Google Scholar] [CrossRef] [PubMed]
- Bisle, S.; Klingenbeck, L.; Borges, V.; Sobotta, K.; Schulze-Luehrmann, J.; Menge, C.; Heydel, C.; Gomes, J.P.; Lührmann, A. The Inhibition of the Apoptosis Pathway by the Coxiella burnetii Effector Protein CaeA Requires the EK Repetition Motif, but Is Independent of Survivin. Virulence 2016, 7, 400–412. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Li, R.; Hu, R.; Deng, X.; Xu, Y.; Zheng, W.; Yi, J.; Wang, Y.; Chen, C. Brucella abortus BspJ Is a Nucleomodulin That Inhibits Macrophage Apoptosis and Promotes Intracellular Survival of Brucella. Front. Microbiol. 2020, 11, 599205. [Google Scholar] [CrossRef]
- Liu, H.; Bao, W.; Lin, M.; Niu, H.; Rikihisa, Y. Ehrlichia Type IV Secretion Effector ECH0825 Is Translocated to Mitochondria and Curbs ROS and Apoptosis by Upregulating Host MnSOD. Cell. Microbiol. 2012, 14, 1037–1050. [Google Scholar] [CrossRef] [Green Version]
- Niu, H.; Kozjak-Pavlovic, V.; Rudel, T.; Rikihisa, Y. Ats-1 Is Imported into Host Cell Mitochondria and Interferes with Apoptosis Induction. PLoS Pathog. 2010, 6, e1000774. [Google Scholar] [CrossRef]
- Schmid, M.C.; Scheidegger, F.; Dehio, M.; Balmelle-Devaux, N.; Schulein, R.; Guye, P.; Chennakesava, C.S.; Biedermann, B.; Dehio, C. A Translocated Bacterial Protein Protects Vascular Endothelial Cells from Apoptosis. PLoS Pathog. 2006, 2, e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulliainen, A.T.; Pieles, K.; Brand, C.S.; Hauert, B.; Böhm, A.; Quebatte, M.; Wepf, A.; Gstaiger, M.; Aebersold, R.; Dessauer, C.W.; et al. Bacterial Effector Binds Host Cell Adenylyl Cyclase to Potentiate Gαs-Dependent CAMP Production. Proc. Natl. Acad. Sci. USA 2012, 109, 9581–9586. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bienvenu, A.; Martinez, E.; Bonazzi, M. Undercover Agents of Infection: The Stealth Strategies of T4SS-Equipped Bacterial Pathogens. Toxins 2021, 13, 713. https://doi.org/10.3390/toxins13100713
Bienvenu A, Martinez E, Bonazzi M. Undercover Agents of Infection: The Stealth Strategies of T4SS-Equipped Bacterial Pathogens. Toxins. 2021; 13(10):713. https://doi.org/10.3390/toxins13100713
Chicago/Turabian StyleBienvenu, Arthur, Eric Martinez, and Matteo Bonazzi. 2021. "Undercover Agents of Infection: The Stealth Strategies of T4SS-Equipped Bacterial Pathogens" Toxins 13, no. 10: 713. https://doi.org/10.3390/toxins13100713
APA StyleBienvenu, A., Martinez, E., & Bonazzi, M. (2021). Undercover Agents of Infection: The Stealth Strategies of T4SS-Equipped Bacterial Pathogens. Toxins, 13(10), 713. https://doi.org/10.3390/toxins13100713