1. Introduction
Bacillus cereus produces various virulence factors including phospholipases, hemolysins and enterotoxins [
1,
2]. One of the enterotoxins considered as a causative factor for the
B. cereus associated diarrheal disease is the multicomponent toxin named Hemolysin BL (Hbl). Early studies have identified three individual antigens that were described as a B binding component and two lytic components, namely L
1 and L
2 [
3,
4]. These subunits are encoded in the
hblCDA operon where
hblC relates to the L
2,
hblD to the L
1 and
hblA to the B Component. Further studies revealed a hemolytic activity when all three subunits were present. It was concluded that the resulting hemolysis is particularly characterized by a ring-shaped zone offsite of the center of the applied toxin. This phenomenon was later explained by a diffusion of the subunits out of an area of L
1 excess which inhibits the hemolytic activity [
5,
6,
7]. Even though Hbl does not only show lytic activity but also enterotoxic activity in rabbit ileal loops [
7,
8], the mode of action was mostly studied using hemolytic systems. Initial studies suggested that all subunits can independently bind to erythrocytes and the lysis is thereby caused by a “membrane attack complex” with the formation of a transmembrane pore of 1.2 nm in diameter [
6]. Later studies indicated a sequential binding order of the B Component followed by the lytic component L
1 and L
2 at last [
9]. The formation of soluble B and L
1 as well as L
1 and L
2 complexes could be verified by surface plasmon resonance spectroscopy (SPR), dot blot analysis and enzyme immuno assays (EIA) [
10]. An oligomerization of the B Component into hepta- or even octamers was suggested [
11], but the complete mode of action of the complex formation still has to be further elucidated. The rapid kinetics and the contribution of each individual subunit to the activity of the whole complex were vastly analyzed, indicating that an excess of the binding Component B led to an accelerated pore-formation while an excess of the lytic components, in particular L
1, delayed this response [
12]. Additional studies have shown an increased effect of the Hbl tripartite enterotoxin when sphingomyelinases and phosphatidylcholine phospholipases C from
Bacillus cereus were present [
13]. It is rather difficult to isolate specific toxins from bacterial strains producing a variety of toxins. Further, the recombinant production and purification of bacterial toxins can be very challenging [
10]. Therefore, the synthesis of individual toxin subunits and their detailed analysis within the whole pore-forming complex is of upmost interest. The synthesis of Hbl in prokaryotic systems has been successfully established in previous studies, but protein purification was necessary [
9]. The addition of tags for purification could alter the folding behavior of the protein itself, which could result in inactive proteins or multicomponent proteins might not assemble. Cell-free protein synthesis (CFPS) is an alternative as proteins synthesized in a eukaryotic system can be directly used for functional assessments without the need for purification. CFPS is a time- and cost-efficient way to produce single proteins, glycoproteins, protein-complexes as well as “difficult-to express” proteins such as membrane proteins and toxic proteins in a well-defined model system [
14,
15,
16,
17,
18,
19,
20,
21]. Single proteins can be synthesized and functionally characterized individually as well as in a mixture of different proteins. Limiting factors such as the cell’s viability or membrane barriers can be neglected as a crude cell lysate instead of intact cells is used [
19,
22]. The open system allows for the supplementation of variable components required for the protein’s need or for further analyses. Therefore, the supplementation of radioactively labeled amino acids allows for the precise protein quantification without the need of any specific antibodies. CFPS is not limited to the use of radioactivity but can fluorescently label the protein of interest site-specifically [
14] or randomly [
23]. In this study, we present cell-free protein synthesis as a tool to rapidly and efficiently study multicomponent toxins and analyze the complex formation out of single subunits in soluble and in a putative membrane bound manner. Here, we examine the Hbl tripartite enterotoxin from
Bacillus cereus, which has not been synthesized in a eukaryotic expression system before. Our cell-free system enables the direct application of the toxin, and toxin subunits in cell-based toxicity assays without prior purification steps as no cytotoxic compounds such as endotoxins are present in eukaryotic cell-free systems. CFPS facilitates the application of multiple synthesis conditions offering the possibility to study the single Hbl subunits as well as their co-expressed complex form. Adding the individual subunits in a sequential manner to the synthesis reaction qualifies the cell-free system for the detailed study of subunit interactions.
3. Discussion
Bacillus cereus strains typically express a variety of toxic proteins among them complex enterotoxins, which cause foodborne diarrheal illness. Until today, no strain has been identified only expressing Hemolysin BL (Hbl). In general, strains that express Hbl also contain the second tripartite toxin Nhe [
25,
26]. To study a specific toxin, time-consuming purification steps are mandatory and need to be optimized in order to guarantee a pure toxin when working with bacterial cultures. Optimized purification procedures have been developed for the Hbl complex, but in the case of tripartite toxins the method has to be adapted and individually optimized for all three subunits [
7,
8]. As the coding sequences for the B, L
1 and L
2 subunits were identified in the 1990s [
27,
28], the specific toxin compounds could be overexpressed in vivo in bacterial strains [
9,
10,
12,
29]. Cell-free protein synthesis (CFPS) represents an efficient method to synthesize such toxic proteins. CFPS does not rely on the cell’s viability as a cell lysate is used which provides ideal prerequisites for the synthesis of toxic proteins [
30]. This study describes the cell-free synthesis of the Hbl complex and its individual subunits for the first time. In addition to that, this is the first study to show the synthesis of Hbl in a eukaryotic system. Using a eukaryotic cell-free system resolves the need to purify the synthesized protein prior to functionality testing which speeds up the screening process. In our study, we show that the cell-free synthesis of each individual subunit as well as a coexpression of two and all three subunits of the Hbl tripartite enterotoxin is possible. In addition to these data, each individual subunit as well as the coexpressed ones were analyzed. Hbl proteins were probed in solution as well as when interacting with the microsomal vesicles present in the CHO lysate.
Table S1 summarizes the data acquired for the hemolytic activity of Hbl. The hemolytic activity of Hbl could be characterized in versatile ways using a eukaryotic cell-free system. Coexpression of all three subunits as well as mixing synthesized individual subunits led to hemolytically active enterotoxin complexes. Using a eukaryotic system, we could reproduce previous findings such as the lytic activity when all three subunits are present but no lytic activity when a single subunit is absent. Further, a crescent zone phenomenon can be achieved when the subunits are in close proximity on the blood agar plates which was shown both in our study and in previous work (
Figure 2 and
Figure S1) [
5].
As Hbl is a soluble protein, the majority of active protein is present in the soluble fraction as compared to the MF (
Figure 2a). Prior studies stated that two structures, namely the LPS-Induced TNF-α Factor (LITAF) and the cell death inducing p53 target 1 (CDIP1), are needed for Hbl to target the cell [
31]. These structures are generally expressed in tissues but are thought to play a role in endosomal protein degradation (the Human protein Atlas). A clear localization to the endoplasmic reticulum cannot be stated. This indicates that the microsomal vesicles used in this study do not harbor LITAF and CDIP1 which could lead to lower protein yields in the MF as no protein accumulation can take place. Even though the Hbl protein did not accumulate in a membranous surrounding leading to a lower protein yield in the MF, the microsomal vesicles in the CHO lysate are beneficial for CFPS. When a microsome depleted lysate was compared to a microsome enriched lysate as presented in
Figure 3a, the overall protein yield was reduced without microsomal vesicles. This might be due to the fact that membrane bound ribosomes at the ER derived microsomes are still present and favor protein translation.
Nonetheless, our results indicated that Hbl might partly interact with the vesicles present in the CHO lysate. We analyzed the membrane association to ER-structures. Using a proteinase K digestion we were able to show that the B Component interacts with the ER-based microsomal vesicles present in our eukaryotic cell-free system whilst the other subunits did not (
Figure 3c and
Figure S3). These data add to previous findings that the B Component anchors to the cell and further allows for complex formation [
3,
4] showing that not only cell surfaces can be targeted but any lipid bilayer. Hemolytic activity of the MF was concentration dependent (
Figure 2d). The MF fraction showed hemolytic activity that was not as intense as for the soluble protein (
Figure 2c) which is likely caused by a concentration-dependent activity on blood agar plates. When studying Hbl subunit interactions our study assessed the different molar plasmid concentrations added to the synthesis as well as the different final protein ratios whilst prior work assessed the protein subunits expressed in bacterial cultures [
4,
6,
10,
12]. In general, both the coexpressed Hbl complex and the complex formed when subunits were mixed after the cell-free synthesis were hemolytically active (
Figure 4). Studies describing the interaction of the different subunits stated that if the L
1 subunit was available in excess to the other subunits, the hemolysis was inhibited [
6] and pore-formation was slowed [
12]. Our work showed lytic activity for all different combinations of molar plasmid ratios as well as molar protein ratios for the SN fraction, except the protein ratios 1:10:1 and 1:1:10 (B: L
2:L
1) (
Figure 4 and
Figure S5). This confirms that an excess of L
1 acts inhibitory and it further indicates that also an excess of the L
2 subunit over both the B and L
1 subunit may impede hemolysis. However, when interpreting these data, it has to be considered that in this study the assessment of lytic activity is only a qualitative analysis and quantitative analyses using precise numbers of erythrocytes could predict the slightest hemolytic activities in the future. Further, an incubation on blood agar plates for 24 h might not be suitable to assess a slower pore-formation. An earlier study has shown that an excess of the B Component inhibits the lytic activity of L
1 [
6]. Within our study, the B Component did not limit the lytic activity at high concentrations. When adding the B Component in a membrane associated way, higher concentrations were needed to facilitate the lytic activity (
Figure 4c). Prior studies indicated a sequential binding order of the B Component followed by the lytic component L
1 and L
2 at last [
9]. Moreover, the formation of soluble B and L
1 as well as L
1 and L
2 complexes could be verified by SPR, Dot Blot analysis and EIA [
10]. The data presented in this study might suggest that the B Component was not fully capable to form pre-complexes with L
1 or anchor L
1-L
2 soluble complexes to the erythrocytes in the MF fraction (
Figure 4b,c). Further findings showed that when the binding component was added to erythrocytes before adding the lytic components, lysis occurred while an addition of the binding component after the lytic components did not induce lysis [
5]. When subunits were fused after cell-free synthesis by an incubation step on ice, no inhibition of the lytic activity could be detected regardless of the combination order of Hbl proteins (
Figure 5). Thus, these data indicate that without the presence of erythrocytes the complex formation is not dependent on the sequential binding order. Previous work has shown that the pore-formation of Hbl is similar to Hemolysin E from
E. coli including a putative oligomerization of the B Component into hepta- and octamers [
11]. Another study has proven the formation of complexes between the lytic component L
1 and the binding component as well as between both lytic components in solution [
10].
The detailed analysis of membrane integrity is best suited to characterize the mode of action of a pore-forming protein and therefore might detect pre-pore complexes. A cell-culture based propidium iodide uptake assay indicated cytotoxicity of the Hbl complex (
Figure 6) in accordance to previous data [
12]. The data acquired in this study are summarized in
Table S2. Jessberger et al., showed a concentration dependent cytotoxic activity of Hbl enterotoxin on Vero cells similar to the results presented here. These previous findings also showed that a 50% inhibition rate of the cell viability was present at 0.3 nM Hbl. In comparison to that, cell-free synthesized Hbl showed initial cytotoxic activity at a concentration of 0.1 nM but could specifically be detected at 0.25 nM in the SN fraction (
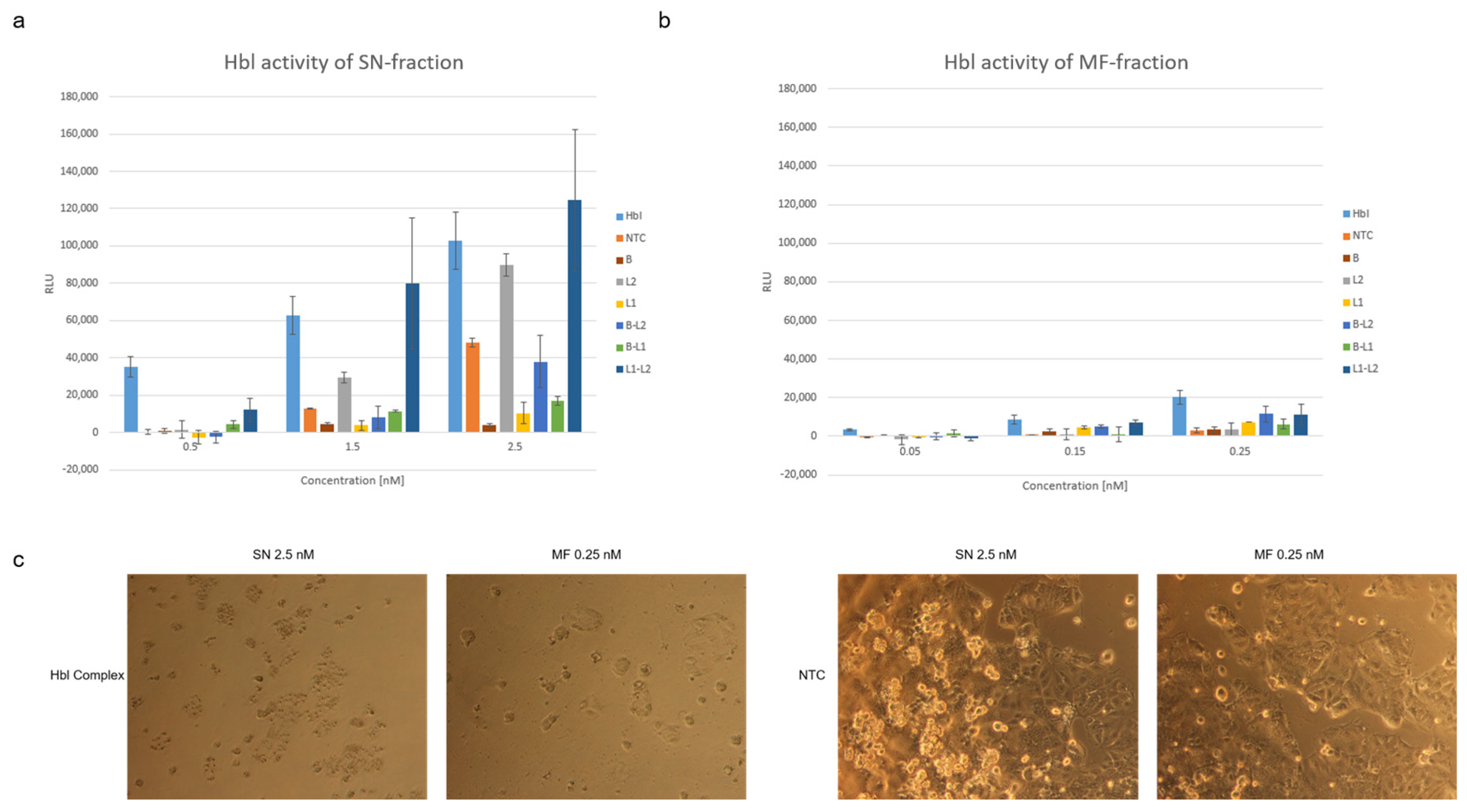
Figure 6a), depicting a similar activity pattern of cell-free synthesized Hbl. Comparing the SN and MF fraction, toxic effects of the MF could already be depicted at concentrations of 0.05 nM (
Figure 6b) as the NTC background measurement presented less intense signals. In contrast to our data, Jessberger et al., further showed that a PI influx was only observed when all three subunits were present during the incubation time on the cells and at an approximate concentration of 3.75 nM no toxic effects for single subunits could be detected [
12]. Our study showed that increasing concentrations of expressed subunits, especially coexpressed subunits, showed high background signals (
Figure 7). These data also depict that the PI uptake assay is applicable to assess cell-free synthesized proteins when lower protein concentrations are applied. At higher target protein concentrations, and when higher volumes of the cell-free reaction were used, background noise could be detected, and further methods should be used such as the MTT viability assay. These background signals might originate from unspecific interactions with the lysate as well as nucleic acids present in the sample mixture. As depicted in
Figure 6, NTC values were higher when using the SN fraction. The overall protein yield was higher in the SN as compared to the MF which led to the use of much higher concentrations of SN protein in the various assays used in this study. This might have led to higher background noise from the lysate as well as the other components added to the synthesis because SN contains a higher fraction of endogenous proteins compared to MF. When assessing the cytotoxic effect of the SN and MF fraction on CaCo2 cells, apoptotic effects were clearly seen for both fractions (
Figure 7) whilst the RLU detected by the PI uptake assay showed higher values for the SN samples. The NTC though did not induce morphological changes, neither when the SN nor when the MF was applied, indicating that the lysate background itself was not cytotoxic. Despite the drawbacks when using the SN fraction, these data demonstrate that the PI uptake assay is an eligible tool to assess the membrane perforation effect. Using low concentrations of cell-free synthesized protein, membrane perforating effects could be detected. This underlines the fact that the PI uptake assay in combination with CFPS can be used for toxicity screenings of pore-forming proteins. The PI uptake assay showed higher RLU values at high concentration for coexpressed L
1-L
2 subunits. Strikingly, they did not show hemolytic activity (
Figure 2). This might be due to the effect that the sample used in this study is not a purified sample. A morphological analysis of CaCo2 cells was performed to assess potential background noise. The data obtained indicate that only the entire Hbl complex induced cell death. In contrast, the single subunits and two coexpressed subunits did not clearly affect the CaCo2 cell viability based on morphological analysis. However, our data also showed a slightly reduced cell confluency when cells were subjected to L
1:L
2 compared to cells subjected to the NTC to untreated cells (
Figure 7c and
Figure S8). These data might indicate a pre-pore-formation of L1 and L2 in a soluble manner interacting with the cell membrane and thereby facilitating the entry of PI into the cell. Such a soluble pre-pore-complex-formation has already been previously described by Tausch et al., 2017, suggesting that cell-free protein synthesis can be applied for the analysis of pre-pore complexes [
10]. A pre-pore formation is often detected in the mechanism of action of pore-forming proteins such as Perfringolysin, ClyA or even Nhe from
B. cereus [
32,
33,
34]. After binding to specific receptors on the cell surface, the pore-forming protein enriches at the targeted cell. With the increased concentration of the protein of interest multimerization and pre-pore-complexes occur. Even though L
1 and L
2 did not show cell binding properties in previous work [
9], Tausch et al., 2017 was the only study yet assessing the detection of the L
1-L
2 complex [
10]. The findings in this study indicate that Hbl-subunits synthesized in a cell-free manner could -assemble to the whole complex or sub-complexes in a soluble as well as a putative membrane associated surrounding. The putative assembly of a pre-pore complex thus has to be further evaluated.
The data acquired in this study qualify CFPS as a promising technology to synthesize and analyze multicomponent proteins in a soluble and membrane associated way. Our results demonstrate that eukaryotic cell-free systems enable the synthesis of a functionally active tripartite Hbl complex via coexpression and when mixing Hbl proteins immediately after their synthesis. In future studies, CFPS might be a valuable tool to study the interaction of Hbl with different toxins from Bacillus cereus such as sphingomyelinases and phosphatidylcholine phospholipases C. In this context, each individual toxin could be synthesized and mixed after cell-free synthesis to study the functional activity of differently combined subunits to resemble the natural conditions. Further in this context, CFPS might help to further elucidate the structural assembly of the Hbl complex. Summarizing, these data depict the significance of CFPS for the synthesis and functional characterization of both soluble and membranous cytotoxic proteins.
4. Material and Methods
4.1. Cell-Free Protein Synthesis
Cell-free synthesis reactions using translationally active lysate derived from Chinese hamster ovary cells (CHO-K1) were performed as described previously [
24,
35,
36]. Shortly, CHO-K1 cells were grown in suspension cultures at 37 °C in controlled fermenters. Cells were cultivated in Power CHO-2 serum free medium (Lonza, Basel, Switzerland) and were harvested at a density of 2 × 10
6 cells/mL. After a centrifugation at 200×
g for 10 min, the cell pellet was washed with 40 mM HEPES-KOH (pH 7.5), 100 mM NaOAc, and 4 mM DTT. Subsequently, cells were disrupted through a micro-emulsifying process using a 20-gauge needle. After a 10 min centrifugation at 10,000×
g, the supernatant was applied to a size-exclusion chromatography column (Sephadex G-25, GE Healthcare, Munich, Germany). Endogenous mRNA was digested by a micrococcal nuclease (S7) treatment for 2 min. The reaction was stopped by adding 6.7 mM EGTA. Lysates were stored at −80 °C until further use. Plasmids encoding each individual subunit of the Hbl enterotoxin were designed according to Brödel et al., 2013 including the internal ribosome entry site (IRES) of the cricket paralysis virus (CrPV) genome to allow for a cap independent translation initiation [
37]. Genes encoding each subunit were obtained by de novo gene synthesis (Biocat GmbH, Heidelberg, Germany). Gene sequences derived from
Bacillus cereus strain ATCC 14,579 were used: AJ237785.1:13463–14590 for the B Component, AJ237785.1:10826–12169 for L
1 and AJ237785.1:12206–13426 for L
2. The coding sequence of the native secretory signal peptide was removed so that only the mature protein was present after cell-free synthesis. All sequences were cloned into the pUC57-1.8K vector backbone and the plasmids were directly used as a template in cell-free reactions. These plasmids were either used separately, in combinations of two plasmids or in a coexpression of all three plasmids to assemble the tripartite toxin. Further, a no template control (NTC) consisting of a translation mixture without any DNA template was used as a background control.
4.2. Batch-Based Reactions
Protein synthesis was conducted in coupled transcription/translation reactions in a final volume of 25 to 80 μL. Batch-based reactions were incubated in a thermomixer (Eppendorf, Wesseling-Berzdorf, Germany) for 3 h at 30 °C and 500 RPM. Cell-free synthesis reactions were composed of 40% (v/v) translationally active CHO lysate supplemented with HEPES-KOH (pH 7.6, f.c. 30 mM, Carl Roth GmbH, Karlsruhe, Germany), sodium acetate (f.c. 100 mM, Merck, Darmstadt, Germany), Mg(OAc)2 (f.c. 3.9 mM, Merck), KOAc (f.c. 150 mM, Merck), amino acids (complete 100 μM, Merck), spermidin (f.c. 0.25 mM; Roche, Grenzach, Germany), Dithiothreitol (DTT, 2.5 mM, Life technologies GmbH, Carlsbad, USA ) and energy regenerating components including creatine phosphokinase (f.c. 0.1 mg/mL, Roche), creatine phosphate (20 mM, Roche), ATP (1.75 mM, Roche) and GTP (0.3 mM, Roche). To allow for DNA transcription during cell-free protein synthesis, 1 U/μL T7 RNA polymerase, 0.3 mM of UTP (Roche) and CTP (Roche) and 0.1 mM of the cap analogue m7G(ppp)G (Prof. Edward Darzynkiewicz, Warsaw University, Warsaw, Poland) were added to the reaction. Additionally, PolyG primer (f.c. 12 µM, IBA, Göttingen, Germany) was supplemented. For further analyses including autoradiography and liquid scintillation counting, cell-free protein synthesis reactions were supplemented with radioactive 14C-leucine (f.c. 50 μM, specific radioactivity 66.67 dpm/pmol, Perkin Elmer, Baesweiler, Germany).
After 3 h of incubation the crude translation mixture (TM) was centrifuged (16,000× g, 10 min, 4 °C) resulting in the supernatant (SN), containing the soluble subunits and the pelleted microsomes containing putative membrane bound subunits. The pellet was resuspended in phosphate buffered saline (PBS) resulting in the microsomal fraction (MF).
4.3. Analysis of Radio-Labeled Proteins
Total protein yields of cell-free synthesized proteins were determined by incorporation of
14C-leucine and subsequent precipitation by hot trichloro acetic acid (TCA, Carl Roth GmbH). Briefly, 3 µL aliquots of the fraction of interest (TM, SN or MF) were mixed with 3 mL of 10% TCA/2% casein hydrolysate (Carl Roth GmbH) solution and incubated at 80 °C for 15 min. After a 30 min incubation on ice,
14C-labeled proteins were transferred to membrane filters (VWR) using a vacuum filtration system (Hoefer, Holliston, USA). Filters were washed twice with 5% TCA to remove non-incorporated
14C-leucine and dried with acetone. Dried filters were placed in 3 mL scintillation cocktail, incubated for at least 1 h and measured by liquid scintillation counting using the LS6500 Multi-Purpose scintillation counter (Beckman Coulter, Germany). The total protein yield of a single protein was calculated according to the following equations:
Total protein yields of de novo synthesized toxin subunits were analyzed using liquid scintillation counting. The total protein yield of coexpressed subunits was estimated using the sum of the molecular weight and the sum of the number of leucines of all expressed subunits.
For qualitative analysis, 3 µL aliquots of the fraction of interest were precipitated in cold acetone (Carl Roth GmbH) and handled as described previously [
38]. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using precast gels (NuPAGE, 10% Bis-Tris, Life technologies) and self-prepared 10% gels using SureCast resolving and stacking buffer (Thermo Fisher Scientific, Rockford, USA ) was performed to confirm the molecular mass of radio-labeled, cell-free synthesized proteins. Precast gels were run at 185 V for 35 min while self-prepared gels were run at 150 V for 55 min. At last, gels were dried at 70 °C (Unigeldryer 3545D, Uniequip, Planegg, Germany), placed on a phosphor screen (GE Healthcare) and radioactively labeled proteins were visualized using a Typhoon Trio and variable mode imager (GE Healthcare).
4.4. Proteinase K Digestion
The protein digestion assay was performed in a final volume of 10 µL. A 5 µL aliquot of the MF was incubated with PBS containing the Proteinase K (NEB) at a final concentration of 1 ng/µL for 30 min on ice. Afterwards the mixture was precipitated with acetone and qualitative analysis was performed as described above.
4.5. Hemolytic Activity Assessment
Hemolytic activity of cell-free produced Hbl enterotoxin was assessed using precast blood agar plates containing 5% sheep blood (VWR). A total volume of 10 µL of de novo synthesized toxin was directly spotted onto the blood agar plate. TM, SN and MF fractions of the cell-free reactions were analyzed. To suppress microbial growth, erythromycin and gentamycin (f.c. 50 µg/mL) were added to each sample. After 24 h of incubation at 37 °C, hemolytic zones were visualized and documented. Triton-X 100 detergent solution was used as a positive control. To analyze defined toxin concentrations, protein yields were initially determined by TCA-precipitation (described in the section entitled “Analysis of Radio-Labeled Proteins”). Subsequently, defined concentrations were calculated, and corresponding volumes were spotted onto the blood agar plate. Experiments were repeated three times.
4.6. Membrane Perforation Studies
In order to assess the insertion of the pore-forming Hbl toxin into the cell membrane, a propidium iodide uptake assay was performed. Therefore, the epithelial colorectal adenocarcinoma cell line CaCo2 (ATCC) was cultured in minimum essential medium (MEM) supplemented with 20% fetal calf serum (Merck, Biochrom GmbH, Berlin, Germany), 1% non-essential amino acids (Merck, Biochrom GmbH) and 1% penicillin/streptomycin (Merck, Biochrom GmbH). In this study, 10,000 CaCo2 cells in 150 µL medium were seeded into 96-well microplates. Cells were allowed to adhere for 24 h and subsequently 20 µL of either the SN or the MF fraction of the cell-free synthesized tripartite enterotoxin were added to each well. Each sample was analyzed in triplicates, if not stated otherwise. As negative controls untreated cells and an NTC synthesized in a batch reaction were used. The latter was diluted and treated in the same manner as the proteinaceous toxin used in the assay and applied in a volume equivalent to the toxin. Toxin concentrations of the radio-labeled SN and MF fractions were measured by TCA-precipitation (see above) and, consequently, assumed for non-labeled fractions as both reactions were prepared in parallel. Varying concentrations from 0.01 nM to 2.5 nM were studied. Additionally, 30 µL of a propidium iodide (MP Biomedicals, Irvine, USA, purchased from VWR, Darmstadt, Germany) solution was added to each well. A final propidium iodide concentration of 10 µg/mL was used. Cells were incubated for 4 h in a cell incubator at 37 °C with 5% CO2. If the membrane of the cell was disrupted by the enterotoxin Hbl, propidium iodide could enter the cell and bind to the DNA. The emission of the propidium iodide was measured at 616 nm using the Mithras LB 943 (Berthold Technologies, Bad Wildbad, Germany) in relative light units (RLU). The mean of each triplicate measurement was calculated and subtracted by the mean triplicate measurement of medium treated cells.
4.7. Morphological Changes of CaCo2 Cells
Morphological changes of CaCo2 cells were analyzed to assess the toxicity of the Hbl enterotoxin. Therefore, cells were seeded into 48 well plates at 15,000 cells/well in 450 µL. Cells were incubated for 24 h to allow for adhesion. Afterwards, the cell-free synthesized toxin was diluted in cell culture medium to the desired concentration and a final volume of 50 µL. These 50 µL were added to the individual wells. Further, 4 h later morphological changes were documented using a light microscope. Phase contrast micrographs were captured with a CCD camera (Nikon, Melville, USA).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






