1. Introduction
Melanoma is the most prominent and lethal skin cancer. The main melanoma mutation is the substitution of a valine to glutamine in codon 600 (V600E) of the serine-threonine kinase BRAF(V600E) which accounts for ~50% of cases [
1]. Melanoma is treatable at early stages by surgical removal. However, advanced or metastatic melanoma becomes lethal.
Breakthroughs have occurred in the treatment of progressed melanoma based on comprehension of the oncogenic signaling, genetic alterations and the immunobiology of this cancer. BRAF inhibitor monotherapy (BRAFi) with the approval of vemurafenib and dabrafenib by the US Food and Drug Administration (FDA) constituted pivotal treatments against melanoma. BRAFi monotherapy has proved to be superior to MEK inhibitor (MEKi) monotherapy, with the latter linked to a narrower therapeutic window [
2]. Unfortunately, in both treatments, patients acquire resistance and eventually relapse [
3]. Drivers of acquired resistance and toxicity of the BRAFi are diverse and include mechanisms leading to paradoxical activation of the mitogen-activated protein kinase (MAPK) pathway [
3,
4]. Interestingly, combining inhibitors of MEK and mutant BRAF kinase delays MAPK-driven acquired resistance and prolongs the duration of responses, achieving higher rate of tumor responses and decreases associated toxicities derived from the paradoxical MAPK pathway activation [
3]. Hence, in advanced BRAF(V600E) melanoma, the standard of care is the combination of BRAF and MEK inhibitors (dabrafenib and trametinib) [
3]. Moreover, the most recently explored option is the triple combination therapy of BRAF and MEK inhibitors with immunotherapy in patients with BRAF(V600E) metastatic melanoma. The combination of BRAFi (dabrafenib) and MEKi (trametinib) with pmel-1 adoptive cell transfer (ACT) causes tumor regression, increases T cell infiltration into tumors and importantly improves in vivo cytotoxicity [
5]. Nevertheless, targeted therapies have been associated with rapid deterioration and death following development of secondary resistance. Hence, there is still the unmet medical need for more efficient targeted approaches against advanced or metastatic BRAF(V600E) melanoma.
In this study we investigated the antitumoral mechanism of action of an octopus-derived peptide (Octpep-1) in BRAF(V600E) melanoma cells. Octpep-1 is a modified version of the previously identified peptide (OCT-TK-III) by the transcriptomics analysis of the
Octopus Kaurna [
6]. We identified the most affected signaling pathways via proteomics and RNAseq analysis and validated the importance of AKT pathway by Western blotting. Finally, we discovered that the combination of Octpep-1 with rapamycin or ERK1/2 inhibitor acts synergistically to potentiate cytotoxicity. These results were coupled with seahorse flux technology to unravel metabolic alterations associated with augmented BRAF(V600E) melanoma cell death caused by the Octpep-1/ERKi combinatory treatment. Altogether, we highlight the therapeutic potential of Octpept-1 in melanoma patients.
3. Discussion
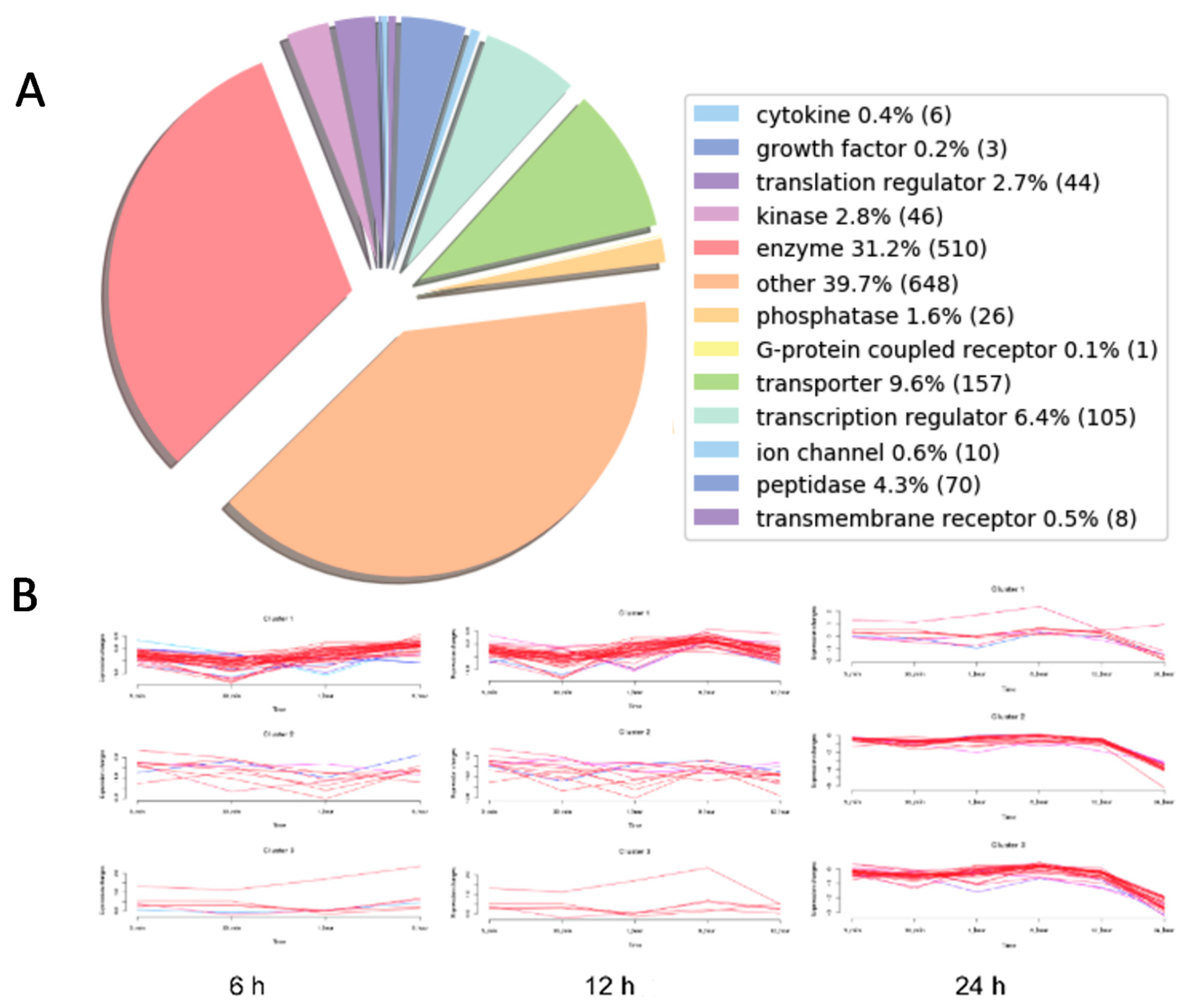
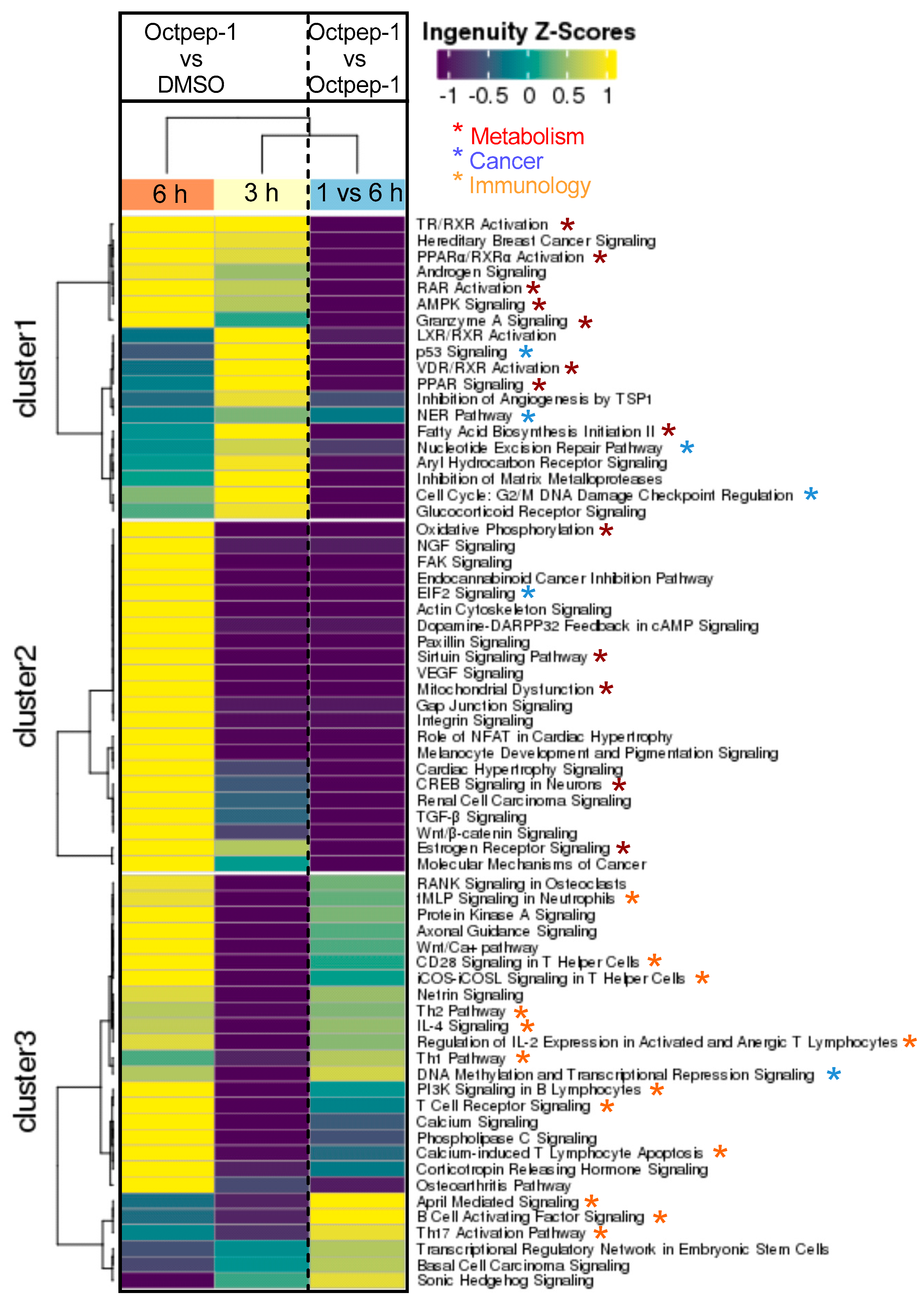
We observed by fluorescence microscopy that Octpep-1 accumulates in MM96L cells to exert its biological activities and this was confirmed by both proteomics and genome-wide gene expression analysis experiments. Proteomics analysis in combination with IPA detailed the clustering of various signaling pathways that were either rapidly or tardily deregulated after Octpep-1 treatment in melanoma cells. Interestingly, there were also pathways such as PI3K/mTOR or actin cytoskeleton that were consistently altered (30 min 24 h).
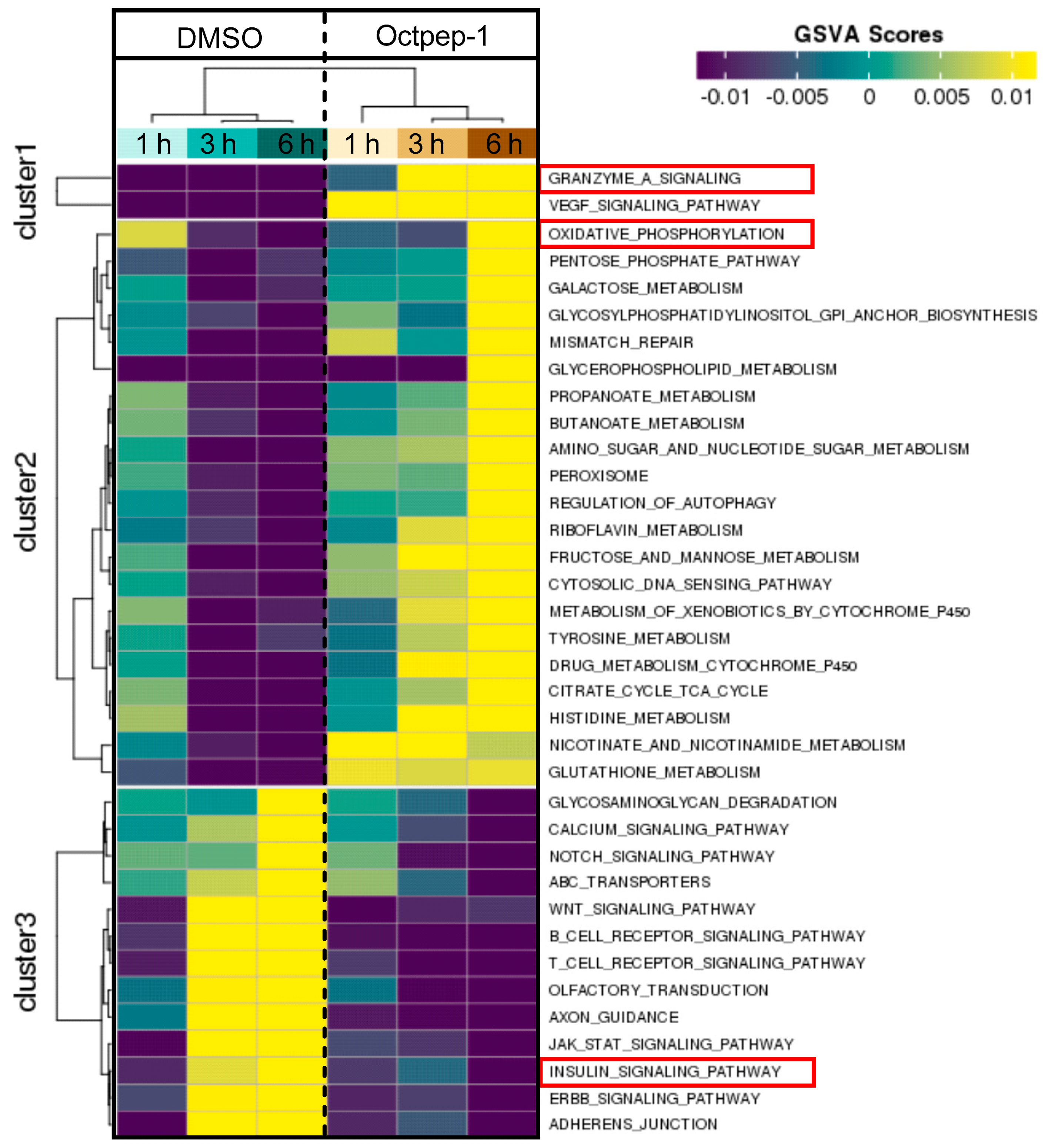
Changes in thioredoxin, granzyme A, and EIF2 signaling pathways suggested that cell survival mechanisms were initiated early within 5 to 30 min of Octpep-1 treatment in MM96L cells. EIF2 activation is of interest since it arrests tumor progression by manipulating the metabolism and redox status [
24]. In addition, the granzyme signaling A pathway activation leads to cell death via caspase-independent pathways [
25]. This was in accordance with lack of cleavage and activation of caspase 3 levels and intact mitochondrial membrane potential after 24 h of octopus peptide exposure (Moral-Sanz et al., unpublished data). After 1h we observed an activation of actin cytoskeleton and VEGF signaling proteins, which are linked to crosstalk and cell migration and thus, to metastasis. Indeed, actin cytoskeleton remained upregulated after 12 h. At this time point, we also noted an enhancement of EIF2 and integrin pathways that most likely contributed to an orchestrated signal transduction cascade related to cell proliferation and metabolism.
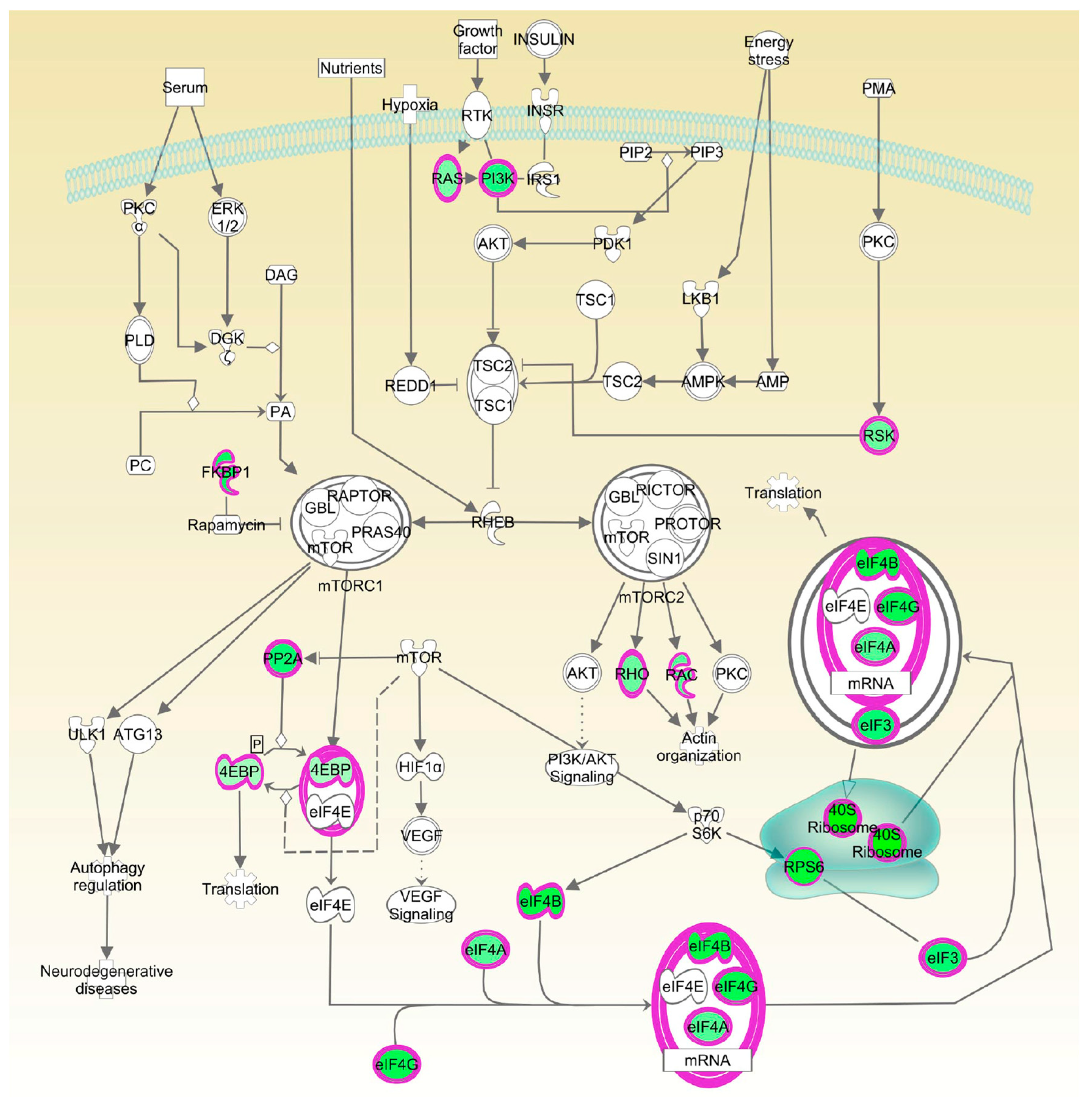
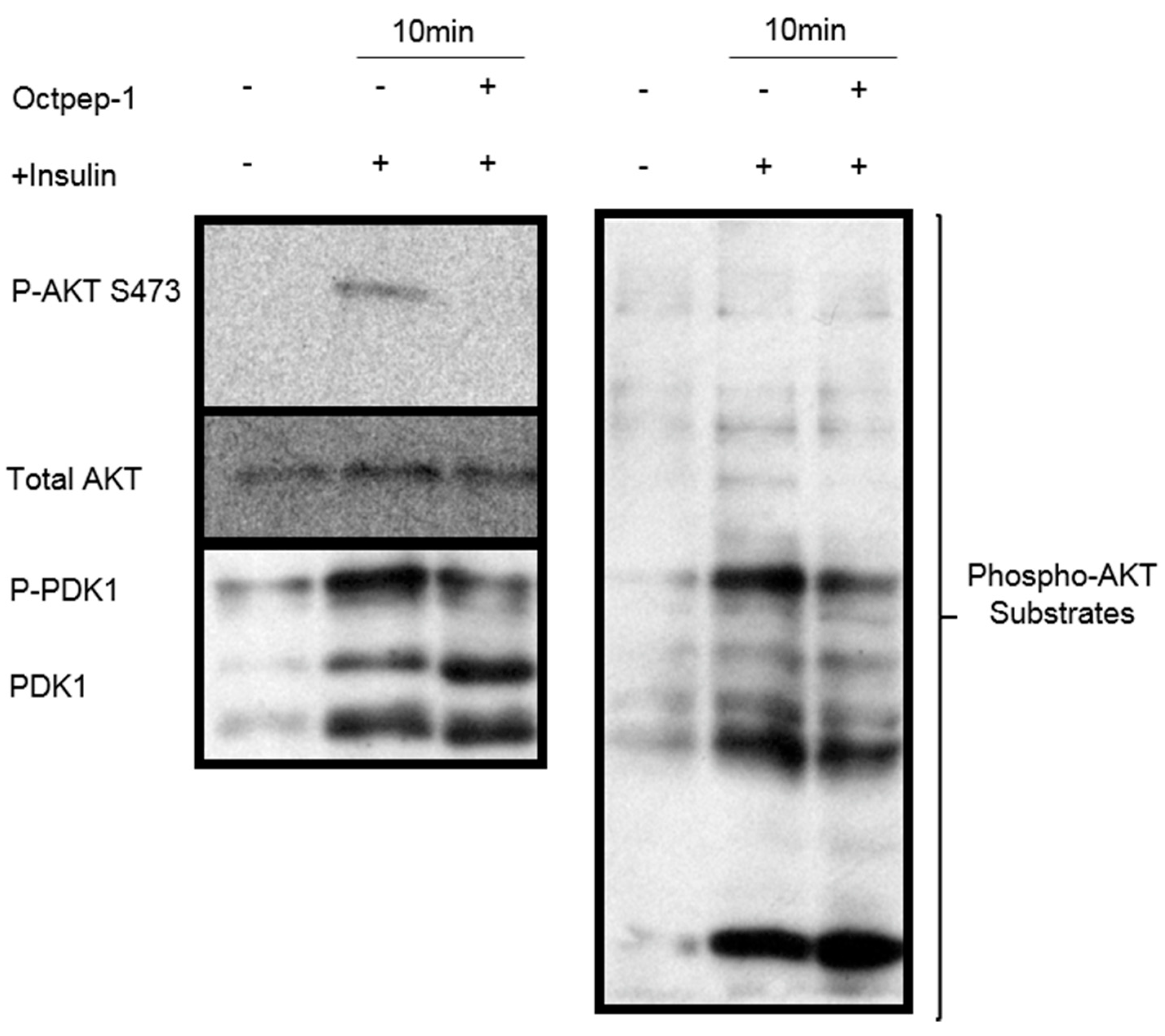
Moreover, the eukaryotic initiation factor-4E (Eif4e)-binding proteins (4E-BPs) are key mTORC1 substrates that control cell proliferation and survival [
26]. The mTOR pathway is located downstream of AKT signaling, with the latter shown to be inhibited by Octpep-1. EIF4B proteins downstream of mTORC1 were also inhibited. This family of proteins leads to the translation of oncogenes and are directly related to tumorigenesis [
27]. Thus, its inhibition most likely prevents proliferation in melanoma cells. Interestingly, upregulated levels of 4E-BPs, which are cancer repressor proteins and mTOR major substrates for phosphorylation [
28] warrant further studies to elucidate a concise description of the mode of action of Octpep-1 alone or in combination with other melanoma therapies.
The proteomics data were supported and complimented further by RNAseq. The fingerprint of Octpep-1 in MM96L cells was grafted by its interactions with granzyme A signaling, VEGF, EIF2, oxidative phosphorylation, insulin signaling, actin cytoskeleton and MMP2 proteins, among others. In addition, RNAseq analysis showcased the downregulation of the ERbB pathway, whose downstream regulators compose the PI3K/AKT/mTOR pathway. We also observed many other metabolic, immunology or cancer-related deregulated signaling pathways, highlighting the wide biological action of Octpep-1 that needs to be unveiled and mapped in melanoma cells.
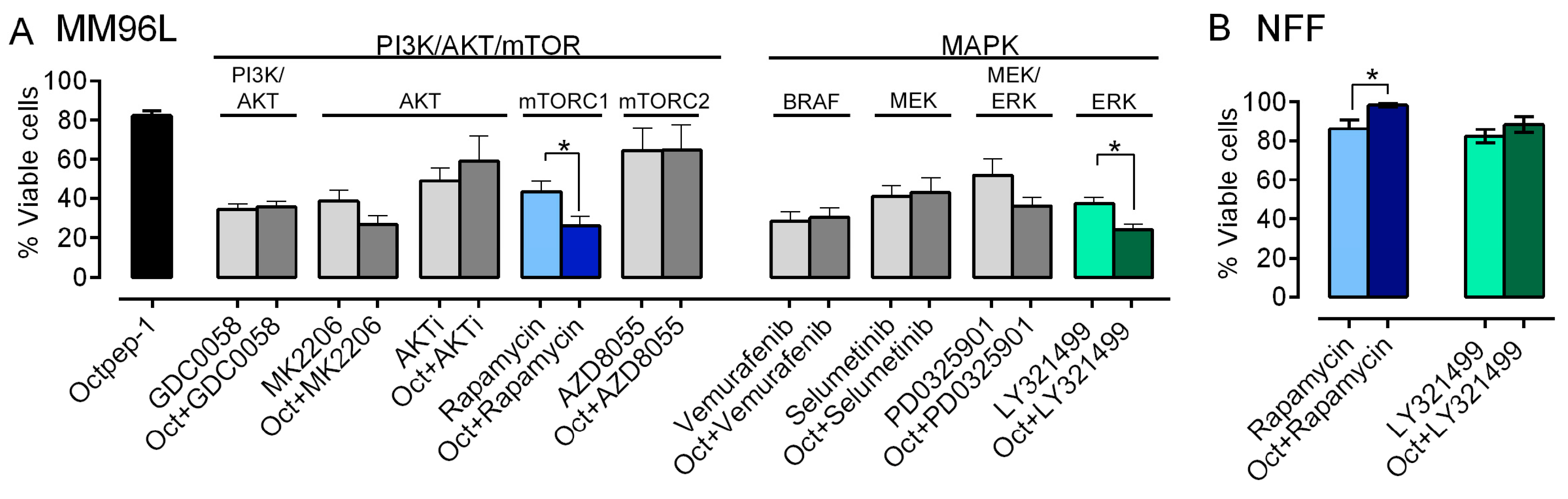
Understanding the mode of action of Octpep-1 is essential to cultivate its therapeutic potential as a drug candidate against melanoma of BRAF-mutation. In the clinic, the rapid antitumoral responses observed with monotherapy inhibitors (BRAFi or MEKi) are undermined due to the development of acquired resistance followed by tumor progression [
3]. This establishes a hallmark in the treatment of advanced or metastatic BRAF-melanoma, which is mainly associated with a paradoxical activation of MAPK [
3,
4]. Hence to circumvent this issue, combinatory approaches are deemed promising. The omics evaluation of our candidate showed that Octpep-1 inhibits the ERbB/PI3K/AKT/mTOR pathway. This prompted us to examine its antiproliferative properties in combination with other inhibitors in melanoma BRAF (V600E) cells. We have characterized in detail the antiproliferative profile of Octpep-1 in melanoma BRAF-mutated cells and have shown that it reduces the tumor progression in nude xenograft mice (Moral-Sanz et al., unpublished data). However, Octpep-1 is active at relatively high concentrations (200 μg/mL in cultured cells vs. 60 mg/kg in mice) (Moral-Sanz et al., unpublished data). Consequently, we examined whether its combination with other inhibitors would be a more effective therapeutic approach against BRAF (V600E) melanoma cells. Indeed, our study revealed specific synergistic antitumoral activities of Octpep-1 in combination with rapamycin (mTOR inhibitor of complex 1) or LY3214996 (EKR1/2i). In both combinations the viability of the melanoma cells was reduced to approximately 24% instead of ≈40% for the inhibitors (rapamycin 5 μM and ERKi 2 μM) and ≈80% for the Octpep-1 (100 μg/mL) alone. Proteomics revealed that 4EBPs that are located downstream of mTORC1 were inhibited. Hence, the blockage of mTORC1 by rapamycin strengthens the effectiveness of our candidate in the blockage of the ERbB/PI3K/AKT/mTOR pathway and potentially arrests the initiation of a drug resistance mechanism in melanoma cells. In addition, the synergistically abolishing effect of Octpep-1 with the ERKi is in accordance with the current literature stating that MAPK is stimulated in BRAF melanoma [
3,
4].
There is an interplay and an extensive crosstalk between MAPK and PI3K/AKT pathways. MAPK resistance to BRAF inhibitors can be mediated through the enhancement of the latter signaling pathway [
29]. In fact, cross-activation of the PI3K/AKT/mTOR pathway is mediated by the activation of ERK or RSK and consequently mTORC1 [
30]. Furthermore, AKT inhibition reverses the acquired drug resistance in combination therapy with dabrafenib and trametinib in vitro [
29]. Synergistic effects and delayed acquisition of resistance have also been shown with the combination of vemurafenib (BRAFi) and SCH722984 (ERKi) in BRAF mutant melanoma cells [
30]. Therefore, it might be a favorable regime to combine a BRAF inhibitor with an inhibitor of the PI3K-AKT like Octpep-1 or MAPK pathway in BRAF melanoma patients.
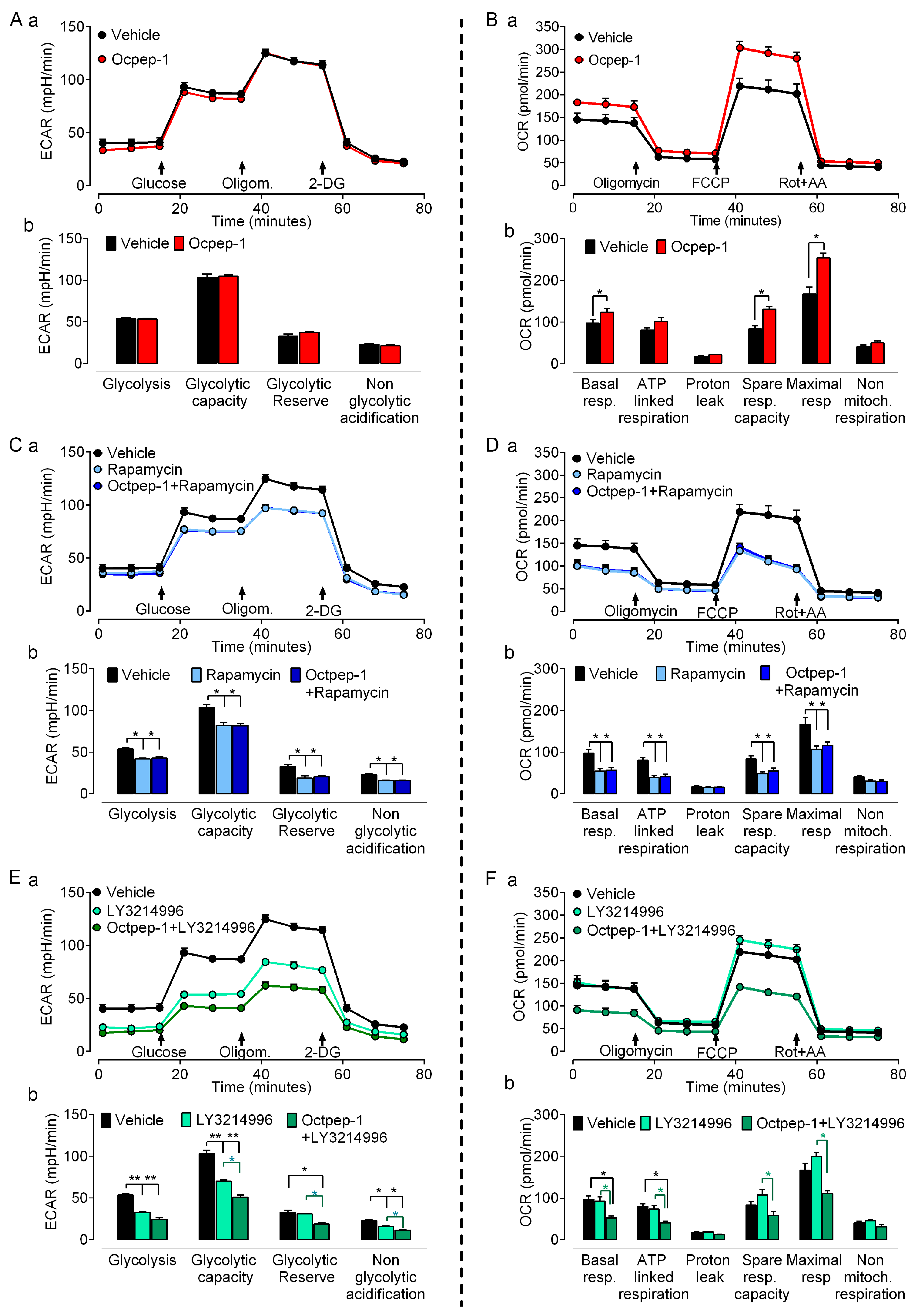
Efficacy of anticancer agents is not solely dependent upon proliferative index. Tumors use diverse mechanisms to thrive and this influences drug effectiveness [
31]. Targeting tumor cell metabolism may either promote or prevent progression and thus could be an attractive anti-tumoral therapeutic approach. In addition, the fact that omics identified various metabolic-related signaling pathways, all together, prompted us to examine the effect of Octpep-1 in various metabolic parameters in melanoma BRAF-mutated cells. The combination of Octpep-1 with the ERK inhibitor coordinated parallel reductions in glycolytic and mitochondrial fluxes that compromised the metabolic flexibility of MM96L cells. Therefore, this challenged metabolism set unfavorable conditions for growth that supports the potential of Octpep-1 to reduce the viability of melanoma cells.
5. Materials and Methods
5.1. Reagents
All media for cell culture, was purchased from Invitrogen/Gibco ThermoFisher, Massachusetts, USA. MitoTEMPO was from Cayman Chemicals (Michigan, USA) (# 16621). Antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA) including: Rabbit anti-Phospho AKTSer473 (#4060), Total AKT (#4691), Phospho-PDK1 Anti-rabbit HRP (#A0545) was from Sigma-Aldrich (Munich, Germany) etc.
5.2. General
All reagents related to chemical synthesis of the compounds were obtained commercially and were used without further purification. Peptides were dissolved in water and mixed 1:1 (v/v) with α-cyano-4-hydroxycinnamic acid matrix (7 mg/mL in 50% MeCN, 5% formic acid) and mass spectra acquired in positive reflector mode. All reported masses are for the monoisotopic [M + H]+ ions. Amino acids were purchased from IRIS Biotech GmbH (Marktredwitz, Germany), Bachem (Bubendorf, Switzerland), or ChemImpex Inc. (Wood Dale, IL, USA). Fmoc-Met-Wang resin was obtained from Peptide International (Louisville, KY, USA).
Eluents for RP-HPLC consisted of 0.05% trifluoroacetic acid (TFA)/H2O (solvent A) and 90% ACN/0.045% TFA/H2O (solvent B). Analytical HPLC quality control analysis of the investigated peptide was performed on a Shimadzu LC20AT system using an Agilent Eclipse Plus C18, 2.1 × 50 mm, 5 μm column with a flow rate of 0.3 mL/min at 45 °C. A gradient of 10 to 50% B over 40 min was used, with detection at 214 nm.
Preparative HPLC were performed on a Waters 600E system using a gradient of 0 to 50% B over 50 min. For the prep. HPLC of the crude peptide an Agilent Eclipse XBD-C18, 21.2 × 250 mm, 5 µM column with a flow rate of 16 mL/min was used. Monoisotopic molecular masses were determined by MALDI-MS on a 4700 Proteomics Analyzer (Applied Biosystems, Waltham, MA, USA) mass spectrometer in positive ion mode.
5.3. Chemical Synthesis of Octpep-1 and Labeled Peptide
Octpep-1 (H-DPPSDDEFVSLM-OH) was assembled on a 0.25-mmol scale on a Fmoc-Met Wang resin following the Fmoc/tBu-SPPS protocol. Couplings were performed in dimethylformamide (DMF) using 4 equivalents of Fmoc-amino acid/
O-(6-Chlorobenzotriazol-1-yl)-N, N, N’, N’-tetramethyluronium hexafluorophosphate (HCTU)/DIPEA (1:1:1.1) relative to resin loading for 15 min. Fmoc deprotection was achieved using 50% piperidine/DMF (2 × 1 min). Final cleavage and global side chain deprotection were accomplished by stirring in 90% TFA, 5% TIPS, and 5% H
2O for 90 min at ambient temperature. The suspension was filtered, washed with TFA, the filtrate concentrated under steady nitrogen flow to a minimal amount, and the peptide precipitated and washed with cold diethyl ether. The precipitate was filtered off and then dissolved in 0.05% TFA in 50% MeCN/H
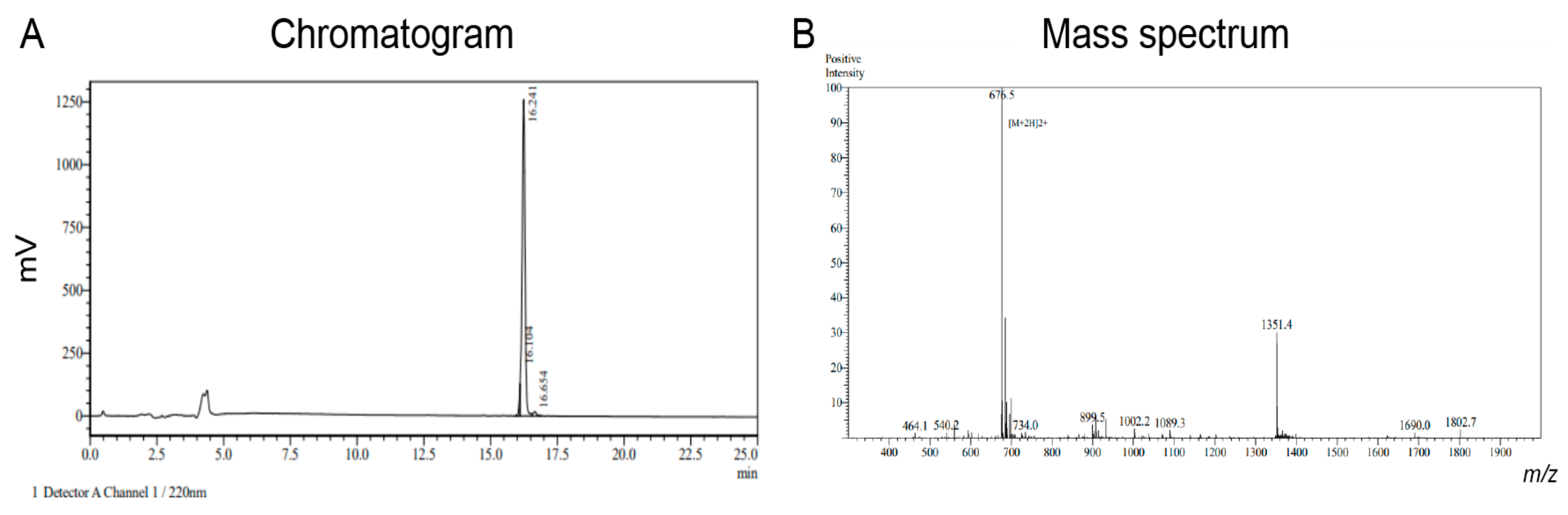
2O and lyophilized. The crude peptide was purified by preparative HPLC and the mass analyzed by MS in positive ion mode using an ABSciex API 2000TM. Pure fractions were combined and lyophilized (6.5 mg) (
Figure A1).
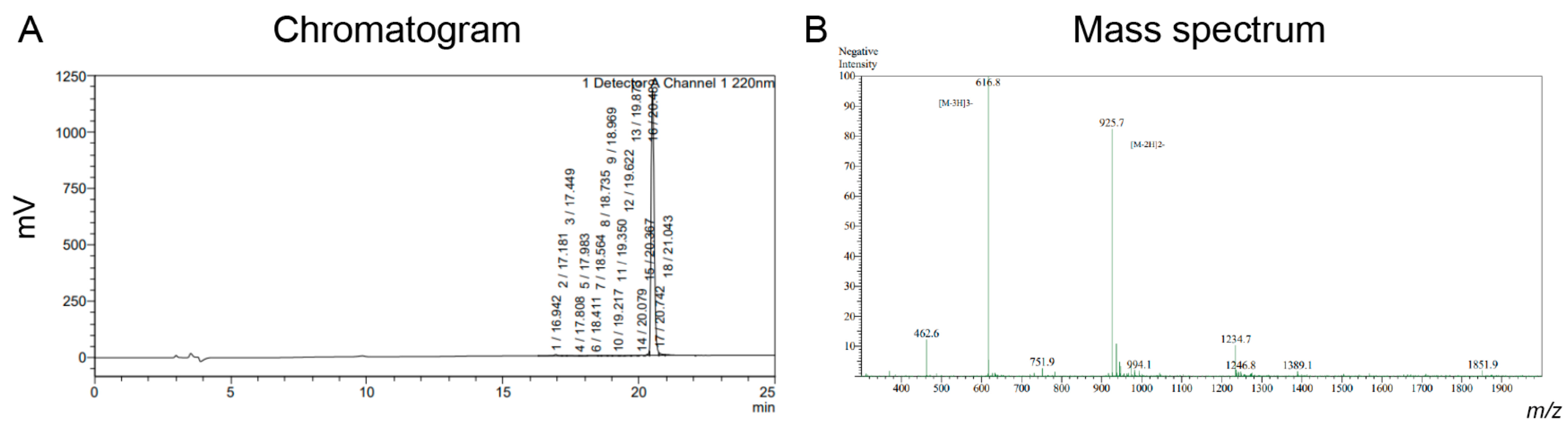
DPPSDDEFVSLM-OH with a modification at N-T:FITC-Ahx was chemically synthesized using FMOC-SPPS and purchased by GenScript. Pure fractions were lyophilized (20 mg) (
Figure A2). In brief, Octpep-1 was tagged with fluorescein isothiocyanate (FITC) and using a spacer, 1,6-aminohexanoic acid (Ahx). For efficient N-terminal labeling of peptides by solid phase synthesis, a spacer is recommended between the fluorophore (fluoroscein) and the N-terminus of the peptide [
32].
5.4. Cell Culture
The MM96L, human melanoma and patient-derived cell line as well as the non-transformed neonatal foreskin fibroblast (NFF) cells were maintained in a humidified incubator at 37 °C and 5% CO2. The melanoma cells were cultured in RPMI-1640 media supplemented with 10% FBS, and 2 mM GlutamaxTM. NFF cells were grown in RPMI-1640 containing 10% FBS. In all cells, penicillin/streptomycin (PS) (100 U/mL each) was added in the media. Cells were passaged at approximately 90% confluency and experiments were performed with passages up to 10. All cell lines were mycoplasma free.
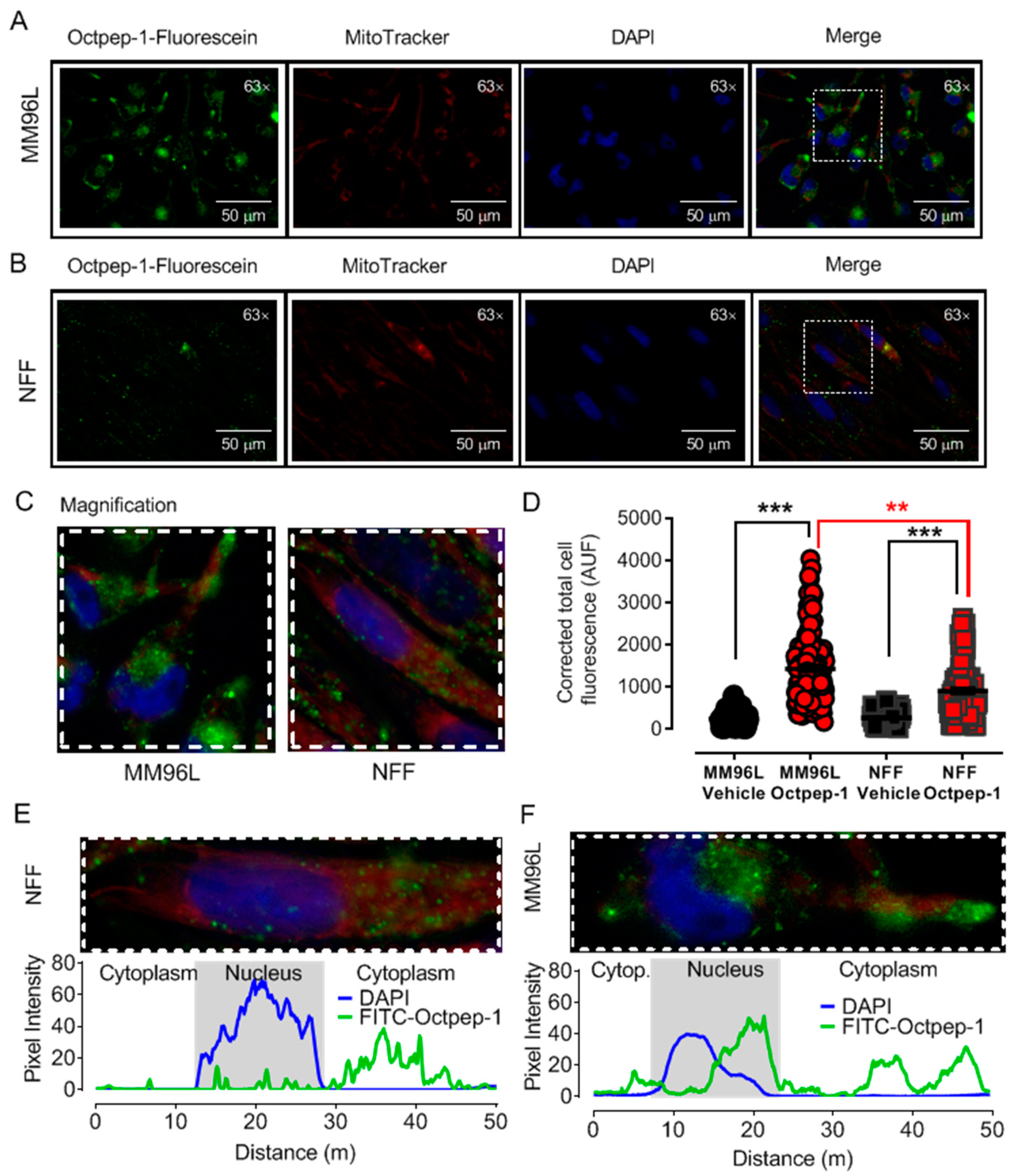
5.5. Fluorescence Microscopy
Octpep-1 tagged with fluorescein at the N-terminus was purchased from GenScript (Leiden, The Netherlands). MM96L cells were grown in coverslips and treated with 200 µg/mL Octpep-1 tagged with fluorescein at 37 °C for 48 h. Additionally, mitochondrial co-staining was performed by incubation with 100 nM MitoTracker Red (excitation, 581 nm; emission, 644 nm) for 30 min at 37 °C followed by washes with PBS and 30 min fixation with 4% PFA at room temperature. Fluoroshield Mounting Medium with DAPI (4′,6-diamidino-2-phenylindole) was used to stain the nucleus and preserve fluorescence in the fixed samples. Images of the emitted fluorescence were acquired at 300, 50, and 100 ms of exposure for green, red, and blue fluorescence, respectively, using a Leica DMIL microscope with a Plan- Apochromat 63 × 1.4 NA oil immersion objective. Analysis was completed using ImageJ-Fiji software (National Institutes of Health, Tennessee, USA). Data are expressed as corrected total cell fluorescence using the following equation [(Cell Area × Mean Cellular Fluorescence) − (Cell Area × Mean Background fluorescence)].
5.6. Cell Lysis of Stimulated and Unstimulated Melanoma Cell Line for Protein Library Generation and SWATH Quantitation
For the protein library generation approximately 30 million near confluent unstimulated adherent MM96L melanoma cells were trypsinised for 2 min and then harvested followed by three washes with Dulbecco’s PBS (Gibco, Waltham, MA, USA) with centrifugation at 800× g for 5 min between each wash. For SWATH quantification, 100,000 adherent mm96l melanoma cells were harvested in triplicate at different time points post-stimulation (unstimulated, 5 min, 30 min, 1, 6, 12, and 24 h) and were washed three times with Dulbecco’s PBS (Gibco, Waltham, MA, USA) with centrifugation at 800× g for 5 min between each wash. Stimulated and unstimulated MM96L cells were lysed on ice by resuspending the cells pellet in 100 mM TEAB (Thriethylammonium bicarbonate), 1% SDS (sodium dodecyl sulfate), 10 mM CHAPS, 5 mM MgCl2 supplemented with 1× Roche Complete protease inhibitors. DNA and RNA were then degraded by the addition of 50 µL and 2 µL, respectively, of 50 mM Tris, 20 mM NaCl, and 2 mM MgCl2 at 1 unit/µl of ultrapure Benzonase (Sigma, Munich, Germany) followed by incubation at 4 °C for 45 min with constant agitation. Protein concentration was determined using the Pierce BCA Assay (ThermoFisher Scientific, Waltham, MA, USA) following the manufacturer’s protocol.
5.7. Peptide Fractionation by OFFGELTM Electrophoresis (OGE) for Protein Library
600 µg of proteins in lysis buffer from the unstimulated MM96L cells were aliquoted and reduced with 0.5 M DTT stock solution added to a final concentration of 20 mM. Samples were then incubated at 75 °C for 10 min and cooled at room temperature for 10 min. Proteins were then alkylated for 30 min in darkness with 0.5 M iodoacetamide (IAA) stock solution to a final concentration of 40 mM. A 10 kDa molecular weight cut-off filter tube (Amicon Ultra-4, Millipore, Burlington, MA, USA) was equilibrated by spinning 700 µL of 100 mM TEAB through the filter at 3100× g for 20 min. The protein sample was mixed with 6 volume of 8 M Urea in 100 mM TEAB, 10% isopropanol, and transferred to the filter unit and spun at 3100× g for 40 min in a 5810R (Eppendorf, Hamburg, Germany) centrifuge. Detergent removal by buffer exchange was performed using a first wash of 500 µL of 8 M Urea in 100 mM TEAB, 10% (v/v) isopropanol followed by a wash with 500 µL of 100 mM TEAB, 10% isopropanol then two successive washes with 500 µL of 50 mM TEAB at 3100× g for 40 min each. The concentrated protein solution was then collected from the filter unit and transferred to an Eppendorf microcentrifuge tube and protein digestion was performed by adding Tryspin enzyme in 50 mM TEAB in an enzyme to protein ratio of 1:50 then mixed well before incubation at 37 °C in a wet chamber overnight. Peptides from the digested protein sample were desalted using a Sep-Pak Vac C18 cartridge (Waters, Milford, MA, USA) and the eluted fraction was lyophilized with a speed vacuum prior to OFFGELTM.
The 3100 OFFGEL Fractionator and OFFGEL kit pH 3-10 (Agilent Technologies, Waldbronn, Germany) with a 24-well setup were prepared according to the manufacturer’s protocols. Lyophilized peptide mixtures were resuspended to a final volume of 3.6 mL using the OFFGEL peptide sample solution. IPG gel strips (24 cm) with a 3–10 linear pH range (GE Healthcare, Munich, Germany) were rehydrated with the Peptide IPG Strip Rehydration Solution according to the manufacturer’s protocol and 150 µL of sample was loaded in each well. Peptides were isoelectrically focused with a maximum current of 50 µA until 50 kV-h were achieved. Twenty-four fractions were recovered from each well and the wells were rinsed with 150 µL of water/methanol/formic acid (49/50/1) for 15 min. Each fraction was lyophilized and then resuspended in 30 µL of H2O with 2% ACN (acetonitrile), 0.1% formic acid (v/v) prior to LC-MS/MS analysis.
5.8. Protein Preparation for SWATH Quantitation
The different protein samples in triplicate for each time point were prepared according to the modified FASP protocol for high throughput proteomics [
33]. Briefly, 50 µg of proteins in lysis buffer from each of the time point replicates were aliquoted and reduced by the addition of 0.5 M DTT stock solution to a final concentration of 20 mM followed by incubation at 75 °C for 10 min and cooling at rt for 10 min. Protein were then alkylated for 30 min in darkness with 0.5 M IAA stock solution to a final concentration of 40 mM. Eight volumes of 8 M Urea, 10% isopropanol in 100 mM TEAB was then added to the protein samples. A 30 kDa molecular weight cut-off filter plate (Acroprep advance 96-well Omega filter plates, PALL, New York, NY, USA), coupled to a deep U-bottom well plate (Axygen, Tewksbury, MA, USA) for collection, was equilibrated by briefly spinning 200 µL of 60% isopropanol through the filter at 3100×
g. Protein samples were then transferred to different wells on the plate and spun at 3100×
g for 30 min in a 5810R (Eppendorf, Hamburg, Germany) centrifuge with adapted plate bucket. Detergent removal by buffer exchange was performed in two successive washes with 8 M urea, 10% isopropanol in 100 mM TEAB with centrifugation at 3100×
g for 30 min between each wash. Urea was then removed by two washes with 10% isopropanol in 50 mM TEAB with centrifugation at 3100×
g for 30 min between each wash. A final wash with 50 mM TEAB was performed with centrifugation as described above. Protein digestion was then performed by adding 1 µg of trypsin in 50 mM TEAB to the wells and incubating overnight at 37 °C. Peptides were recovered over the deep V-bottom plate (Axygen, Life Sciences, Arizona, USA) using an initial spin at 3100×
g for 10 min followed by two centrifugations with 50 µL of 50 mM TEAB. Recovered peptides were dried in a speed vacuum for 4 h at 45 °C. Dried peptides were resolubilized in H
2O, 0.1% TFA (
v/
v), desalted and normalized to 5 µg using ZipTip
TM C18 tips (Millipore, Massachusetts, USA) according to manufacturer’s protocol. Eluted peptides were lyophilized and then resuspended in 30 µL of H
2O with 2% ACN (acetonitrile), 0.1% formic acid (
v/
v) spiked with the iRT calibrant (Biognosys, Zurich, Switzerland) prior to LC-MS/MS analysis.
5.9. Protein Identification/Quantification Using LC-MS/MS
All peptide samples were chromatographically separated by a 10 µL injection on an Eksigent cHiPLCTM-nanoflex system using a 15 cm long chromXP C18-CL column (particle size 3 µM, 120 Å, 200 µM × 6 mm). A pre-concentration step (10 min) was performed employing a chromxp trap (C18-CL, 3 µM, 120 Å, 200 µM × 6 mm) before commencement of the gradient. A flow rate of 500 nl/min was used for all experiments. The mobile phase consisted of solvent A (0.1% formic acid [aq]) and solvent B (100 acetonitrile/0.1% formic acid [aq]) were used for the three consecutive linear gradients for peptide elution: 5–10% solvent B (acetonitrile/0.1% formic acid) over 2 min, 10–40% solvent B over 58 min and 40–50% solvent B over 5 min. A final gradient from 50% to 95% solvent B in 10 min was used to clean the column. Eluates from the RP-HPLC column were directly introduced into the NanoSpray II ionisation source of a TripleTOF 5600 MS/MS System (AB Sciex, Framingham, MA, USA) operated in positive ion electrospray mode. The data for OFFGEL fractions were obtained using Data Dependent Acquisition (DDA) as previously described [
34], while the quantification data from the different time points were obtained using the Sequential Windowed Acquisition of All Theoretical Fragment Ion Mass Spectra method (SWATH) under the same conditions as the DDA experiments and a rolling collision energy method was used to fragment all ions in a set of 26 sequential overlapping windows of 25 AMU over a mass range coverage of 350–1000 (
m/
z). An accumulation time of 100 ms was used for each fragment ion scan resulting in a total cycle time of 2.9 s. Data were acquired and processed using Analyst TF 1.7 software (AB SCIEX, Victoria, Australia).
5.10. Protein Library Generation and Bioinformatic Analysis of SWATH Protein Quantitation
Spectral searches of processed LC-MS/MS data were performed using ProteinPilot v4.5 (AB SCIEX) using the Paragon algorithm (version 4.5.0.0). Background correction was used and biological modifications specified as an ID focus. The detected protein threshold was set as 0.5 and the false-discovery rate (FDR) was calculated using searches against a decoy database comprised of reversed sequences. Searches were conducted against the UniProt human reference proteome set comprising 70,953 protein sequences (downloaded 13th February 2017). For spectral library generation and SWATH XIC peak area extraction PeakView v2.2.0 (AB SCIEX, Victoria, Australia) with the SWATH acquisition MicroApp was used with ion library parameters set to 6 peptides per protein, 6 transitions per peptides, a peptide confidence threshold of 99%, and FDR threshold to 1%. The XIC time window was set to 6 min and XIC width to 75 ppm. Retention times for all SWATH experiments were normalized using the spiked iRT calibrants. To generate the quantitation table files for ions, peptides, and proteins Marker View v1.2.1.1 (AB SCIEX, Victoria, Australia) was used and the relative area under peaks across the different experiments was normalized based on the iRT internal calibrant. Figures and statistical analysis were generated with Python and R language and graphical packages.
5.11. Western Blots
MM96L cells were lysed in cold RIPA buffer containing protease (Merck Pty Ltd., Kilsyth, Australia) and phosphatase (Roche Diagnostics, Castle Hill, Australia) inhibitors and stored at −20 °C. Protein concentrations were determined using a Pierce BCA Protein assay kit (Thermo Fisher Scientific, Waltham, USA). Samples were subjected to SDS-PAGE and blotted according to standard procedures. In brief, 10 μg of protein was loaded per lane. Antibodies used for Western blots are described in the Reagents section. Protein signals were visualized using enhanced chemiluminescence (Pierce™ ECL Western Blotting Substrate, Thermo Fisher Scientific, Waltham, MA, USA).
5.12. RNA Sequencing
Time course was performed at 30 min, 1 h, 3 h, and 6 h of MM96L cells treated with Octpep-1 at 200 μg/mL. The cells were then collected, and RNA extraction was performed by RNAsy kit (QIAGEN, Venlo, The Netherlands) according to manufacturer’s directions. The RNA samples were sent for sequencing to the Institute for Molecular Bioscience Sequencing Facility.
5.13. Pathway Analysis
To determine the differentially expressed set of genes for melanoma treated with the Octpep-1 versus vehicle treated cells at different time points, we performed Ingenuity Pathway Analysis (IPA) followed by Principal Component Analysis (PCA) and enriched with GSVA and Ingenuity Z score analysis.
5.14. Cell Viability Assay
Cell viability was evaluated by the MTT assay and measured at 570 nm absorbance on a microplate reader (BIOTEK PowerWave XS, Winooski, VT, USA) after 48 h of peptides’ treatment using an MTT assay (Sigma-Aldrich (Munich, Germany) as previously described [
11,
35].
5.15. Bioenergetics
Cellular bioenergetic measurements were performed using the Seahorse XFe96 Analyzer and XFe96 culture microplates (Agilent Technologies, Madrid, Spain) to investigate Oxygen Consumption Rates (OCR) and Extracellular Acidification Rates (ECAR) in MM96L. Measurements were performed after 24 h incubation with Octpep-1, ERK1/2i, mTORC1i and combination of the inhibitors with Octpep-1 in 15 × 103 melanoma cells/well in quintuplicates. OCR was tested in Seahorse XF base medium containing 15 mM glucose, 1 mM sodium pyruvate, and 2 mM L-glutamine (pH 7.4) and ECAR in Seahorse XF base medium containing 0.5 mM sodium pyruvate and 1 mM L-glutamine (pH 7.4).
5.16. Statistical Analysis
Statistical analyses were conducted using Prism version 8.0 (Graphpad Software Inc., San Diego, CA, USA). Data were checked for normality to choose the appropriate test (parametric or non-parametric test). Significance for the screening of various inhibitors and their combination with Octpep-1 was calculated using a one-way ANOVA and Sidak’s multiple comparisons test. Differences between Octpep-1 and its combination with rapamycin or ERKi were examined by unpaired two-tailed t-test and Mann Whitney test, respectively. In addition, for the seahorse metabolic experiments significance was calculated using a one-way ANOVA and Dunn’s multiple comparison test with p-values adjusted using Kruskal–Wallis or Mann–Whitney method. The data from MTT experiments are the result of at least three independent experiments done in triplicate or quadruplicate while seahorse data are the outcome of four to six independent experiments done at least in quadruplicate. Data are represented as mean ± SEM. Significance was defined at * p ≤ 0.05 or ** p ≤ 0.01.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









