Variability in the Occupancy of Escherichia coli O157 Integration Sites by Shiga Toxin-Encoding Prophages

 ,
,
Abstract
:1. Introduction
2. Results
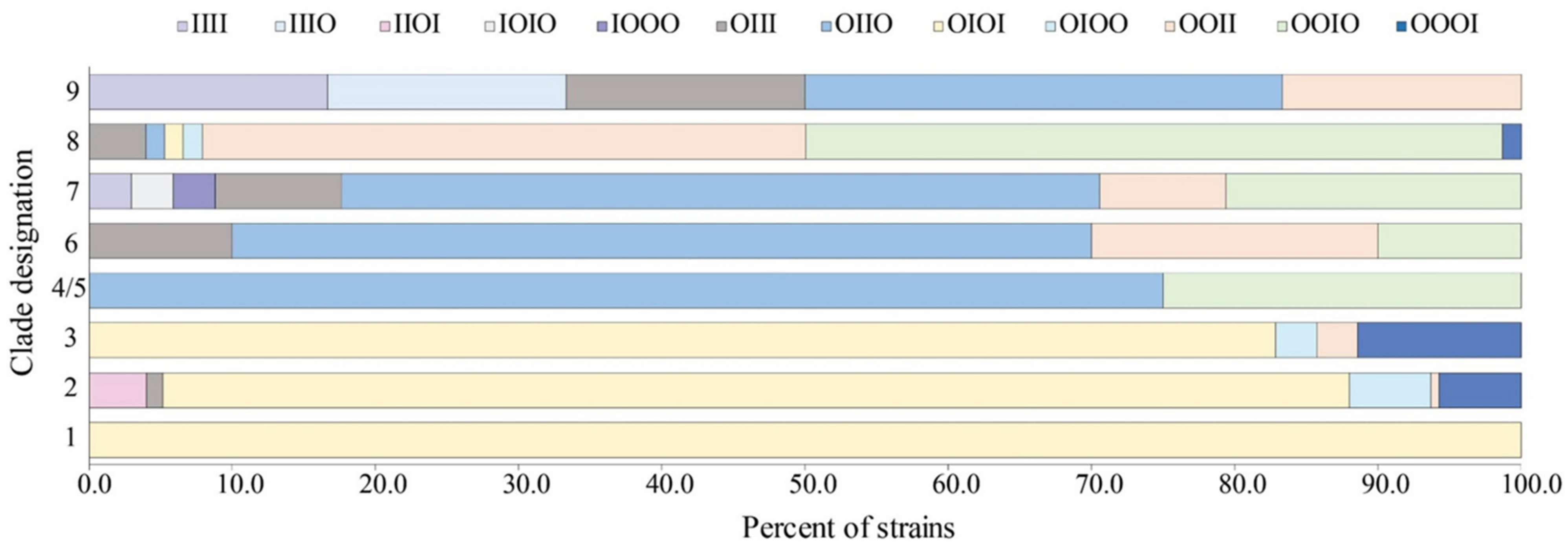
2.1. Prophage Occupancy Varied between the Four Insertion Sites and Differed by Clade
2.2. Specific Prophage Occupancy Profiles Predominated in O157 Strains
2.3. Discrepancies Identified between Occupancy Profiles and the Presence of Stx Variants
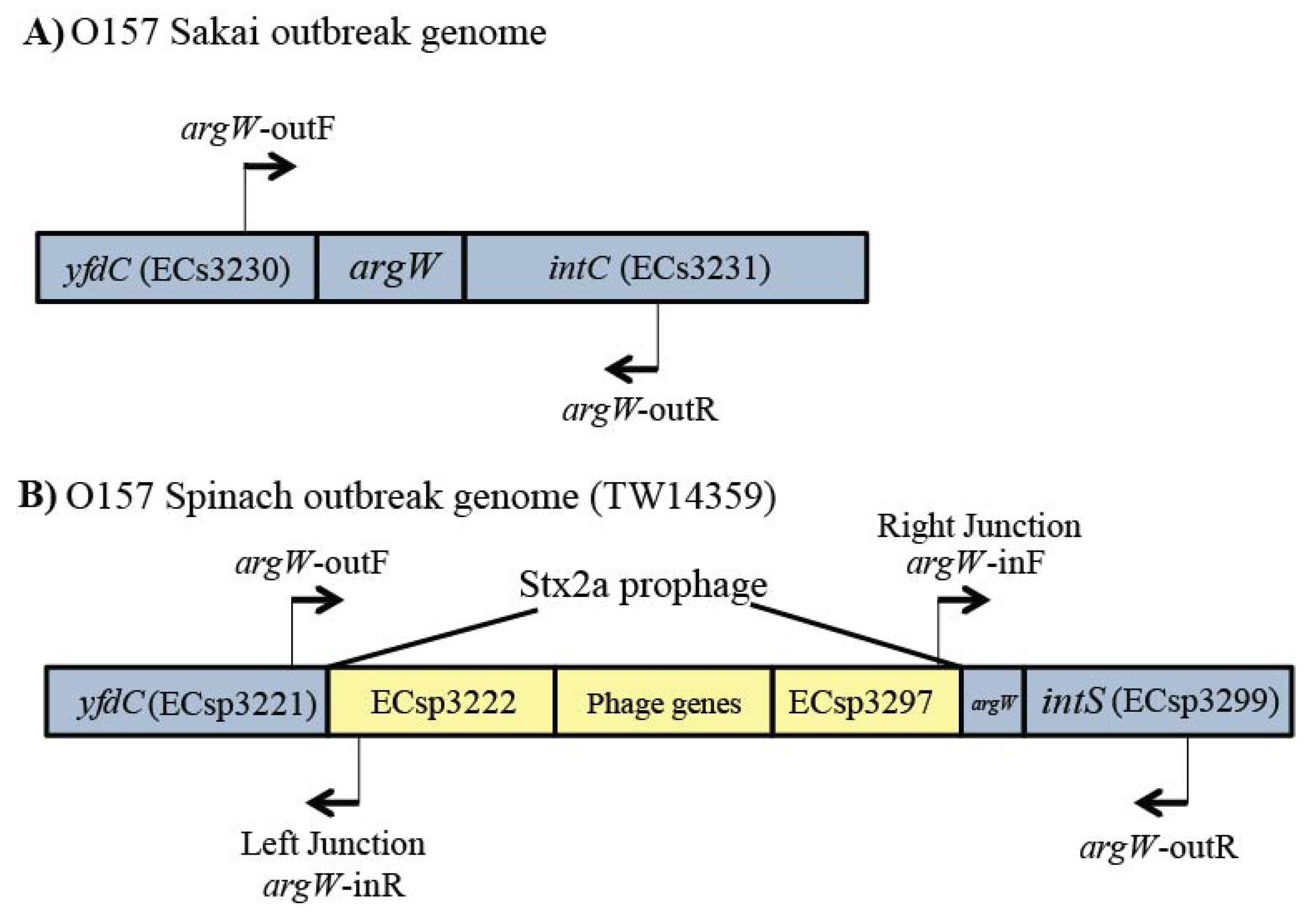
2.4. Confirmation of Stx-Encoding Prophages at Specific Occupied Sites
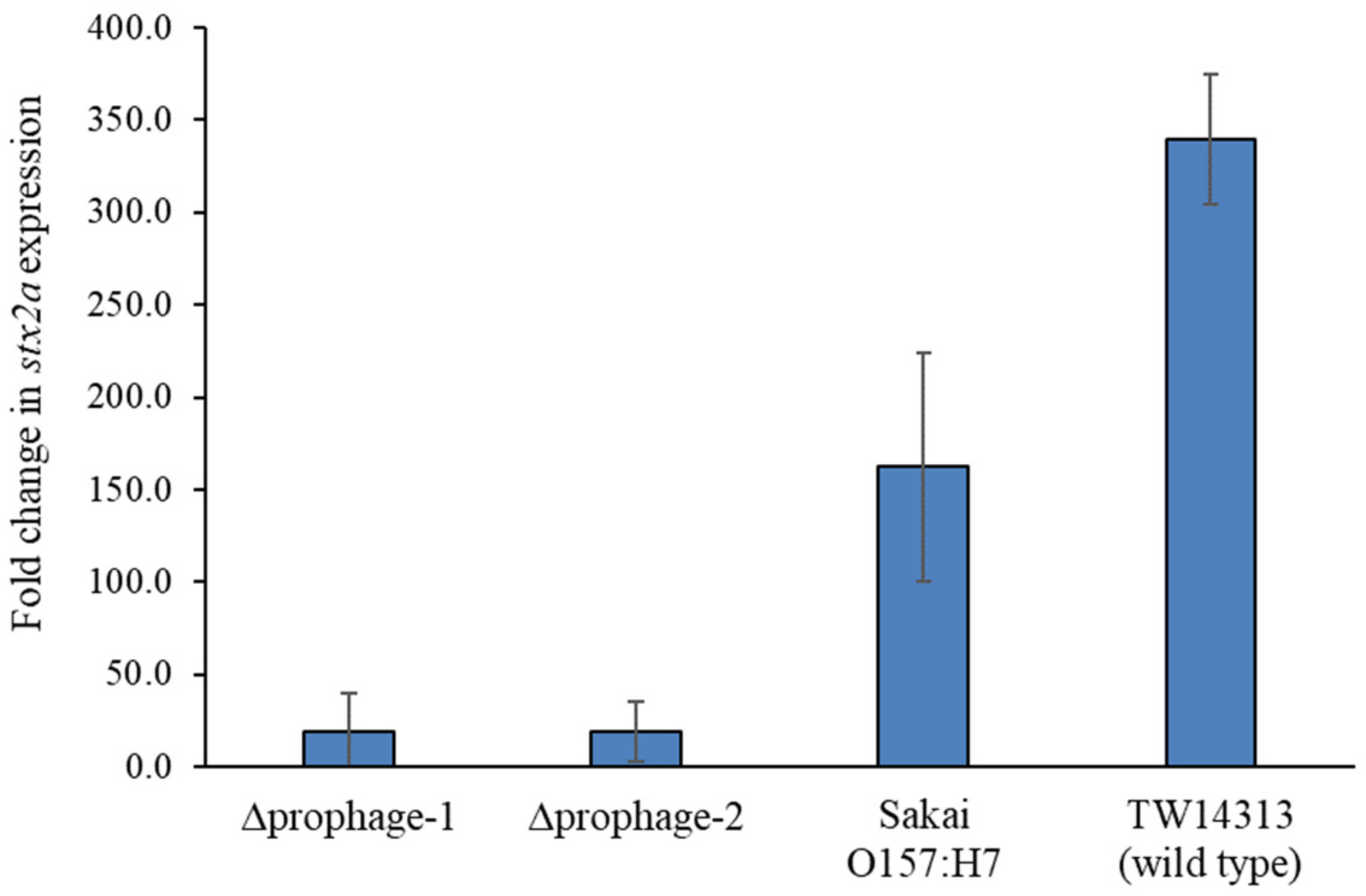
2.5. Non-Stx-Encoding Prophages Could Affect stx2a Expression
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains
4.2. PCR Assays to Detect Prophage Integration
4.3. Frequency of Prophage Occupancy Profiles
4.4. Long-Range PCR to Confirm Insertion Site Occupancy with Stx Prophages
4.5. Deletion of a 20 kb Non-Stx Prophage in TW14313
4.6. Quantifying stx2a Expression
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Product Target | Primer Name | Primer for Sakai | Primer for TW14359 1 | Sequence 5′ to 3′ | Size |
|---|---|---|---|---|---|---|
| yehV | Intact yehV | yehV-outF | ECs2937 | ECSP2931 | GAAGCAGCAGCAACACCAGATTA | 543 bp |
| yehV-outR | ECs3014 | ECSP2998 | ATTGCCGCTTTGCAGGTAGGT | |||
| yehV left junction | yehV-outF | 607 bp | ||||
| yehV-inR | ECs2939 | ECSP2932 | ACAGCACCGTTTGGCATTTTAG | |||
| yehV right junction | yehV-inF | ECs3013 | ECSP2997 | GAAGCAGAGGGAATATGGGAGAACT | 654 bp | |
| yehV-outR | ||||||
| yehV with Stx1a phage | yehVlong-F | ECs2974 (stx1a) | Absent | CAAACAAATTATCCCCTGTGCCACTA | 19.4 kb | |
| yehVlong-R | ECs3015 (yehW) | ECSP3000 (yehW) | AGTTGCGAAAGGTATGGGAATGAGTC | |||
| argW | Intact argW | argW-outF | ECs3230 | ECSP3221 | CTGGCCCTTCGCACTACCTACTT | 527 bp |
| argW-outR | ECs3231 | ECSP3299 | TCTTTCGGCTTTGCTGCTTCA | |||
| argW left junction | argW-outF | 442 bp | ||||
| argW-inR | Absent | ECSP3222 | GGATCGGTTTAACCGCGAGTAC | |||
| argW right junction | argW-inF | absent | ECSP3297 | TTAGCCGACGAACGATCAATAGGT | 1395 bp | |
| argW-outR | ||||||
| argW with Stx2a phage | argWlong-F | Absent | ECSP3253 (stx2a) | GAGGGGTCGATATCTCTGTTCGTATACTATTTA | 25.6 kb | |
| argWlong-R | ECs3231 (intC) | ECSP3299 (intS) | ATTTCAAGGCCGCCGATGATAGGT | |||
| wrbA | Intact wrbA | wrbA-outF | ECs1159.5 2 | ECSP1174 | TACCGCCGCGAACCTGTG | 1118 bp |
| wrbA-outR | ECs1252 | ECSP1175 | CTTTGGCGCGATTTTACTCAATG | |||
| wrbA left junction | wrbA-outF | 605 bp | ||||
| wrbA-inR | ECs1160 | Absent | CCAGCGCCAGCATGGTCTAC | |||
| wrbA right junction | wrbA-inF1 | ECs1251 | Absent | TTTTTCCCTCGCCCATAACCTAT | 1445 bp | |
| wrbA-outR | ||||||
| wrbA with Stx2a phage | wrbAlong-F | ECs1158 (agp) | ECSP1172 (agp) | GAAAGTCGGCAACTCGCTGGTAGA | 22.4 kb | |
| wrbAlong-R | ECs1205 (stx2A) | Absent | AAGTCTATCGTAAACTCCCGGGAATA | |||
| sbcB | Intact sbcB | sbcB-outF | ECs2812 | ECSP2683 | TGACCGGTGGTGTAATCCATCA | 636 bp |
| sbcB-outR | ECs2813 | ECSP2762 | CGGTACGGTAAAAAGCGAGTGAAT | |||
| sbcB left junction | sbcB-outF | 1441 bp | ||||
| sbcB-inR | Absent | ECSP2685 | GTTTGTGATTTCGCGCTGTAGGT | |||
| sbcB right junction | sbcB-inF | Absent | ECSP2761 | CGCTTTGCGCTGAGCCTCTAT | 1588 bp | |
| sbcB-outR | ||||||
| sbcB with Stx2c phage | sbcBlong-F | Absent | ECSP2722 (stx2cB) | CGGCGCCATTGCATTAACAGAAACTA | 26.3 kb | |
| sbcBlong-R | ECs2815 (yeeE) | ECSP2764 (yeeE) | TTGCGTTAATTCAGGCGGGCCTACT |
References
- Karmali, M.A.; Petric, M.; Lim, C.; McKeough, P.C.; Arbus, G.S.; Lior, H. The association between idiopathic hemolytic uremic syndrome and infection by verotoxin-producing Escherichia coli. J. Infect. Dis. 1985, 151, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Scallan, E.; Hoekstra, R.M.; Angulo, F.J.; Tauxe, R.V.; Widdowson, M.A.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne illness acquired in the United States-Major pathogens. Emerg. Infect. Dis. 2011, 17, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Gould, L.H.; Mody, R.K.; Ong, K.L.; Clogher, P.; Cronquist, A.B.; Garman, K.N.; Lathrop, S.; Medus, C.; Spina, N.L.; Webb, T.H.; et al. Increased recognition of Non-O157 Shiga toxin-producing Escherichia coli infections in the United States during 2000–2010: Epidemiologic features and comparison with E. coli O157 infections. Foodborne Pathog. Dis. 2013, 10, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Tseng, M.; Sha, Q.; Rudrik, J.T.; Collins, J.; Henderson, T.; Funk, J.A.; Manning, S.D. Increasing incidence of non-O157 Shiga toxin-producing Escherichia coli (STEC) in Michigan and association with clinical illness. Epidemiol. Infect. 2016, 144, 1394–1405. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, A.D.; Newland, J.W.; Miller, S.F.; Holmes, R.K.; Smith, H.W.; Formal, S.B. Shiga-like toxin-converting phages from Escherichia coli strains that cause hemorrhagic colitis or infantile diarrhea. Science 1984, 226, 694–696. [Google Scholar] [CrossRef]
- Melton-Celsa, A.R. Shiga toxin (Stx) classification, structure, and function. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [Green Version]
- Ostroff, S.M.; Tarr, P.I.; Neill, M.A.; Lewis, J.H.; Hargrett-Bean, N.; Kobayashi, J.M. Toxin genotypes and plasmid profiles as determinants of systemic sequelae in Escherichia coli O157:H7 infections. J. Infect. Dis. 1989, 160, 994–998. [Google Scholar] [CrossRef]
- Boerlin, P.; McEwen, S.A.; Boerlin-Petzold, F.; Wilson, J.B.; Johnson, R.P.; Gyles, C.L. Associations between virulence factors of Shiga toxin-producing Escherichia coli and disease in humans. J. Clin. Microbiol. 1999, 37, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, A.W.; Bielaszewska, M.; Zhang, W.L.; Pulz, M.; Kuczius, T.; Ammon, A.; Karch, H. Escherichia coli harboring Shiga toxin 2 gene variants: Frequency and association with clinical symptoms. J. Infect. Dis. 2002, 185, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Orth, D.; Grif, K.; Khan, A.B.; Naim, A.; Dierich, M.P.; Würzner, R. The Shiga toxin genotype rather than the amount of Shiga toxin or the cytotoxicity of Shiga toxin in vitro correlates with the appearance of the hemolytic uremic syndrome. Diagn. Microbiol. Infect. Dis. 2007, 59, 235–242. [Google Scholar] [CrossRef]
- Persson, S.; Olsen, K.E.P.; Ethelberg, S.; Scheutz, F. Subtyping method for Escherichia coli Shiga toxin (Verocytotoxin) 2 variants and correlations to clinical manifestations. J. Clin. Microbiol. 2007, 45, 2020–2024. [Google Scholar] [CrossRef] [Green Version]
- Louise, C.B.; Obrig, T.G. Specific interaction of Escherichia coli O157:H7-derived Shiga-like toxin II with human renal endothelial cells. J. Infect. Dis. 1995, 172, 1397–1401. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; McAteer, S.P.; Tree, J.J.; Shaw, D.J.; Wolfson, E.B.K.; Beatson, S.A.; Roe, A.J.; Allison, L.J.; Chase-Topping, M.E.; Mahajan, A.; et al. Lysogeny with Shiga toxin 2-encoding bacteriophages represses type III secretion in enterohemorrhagic Escherichia coli. PLoS Pathog. 2012, 8, e1002672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohnishi, M.; Terajima, J.; Kurokawa, K.; Nakayama, K.; Murata, T.; Tamura, K.; Ogura, Y.; Watanabe, H.; Hayashi, T. Genomic diversity of enterohemorrhagic Escherichia coli O157 revealed by whole genome PCR scanning. Proc. Natl. Acad. Sci. USA 2002, 99, 17043–17048. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, N.; Tarr, P.I. Escherichia coli O157:H7 Shiga toxin-encoding bacteriophages: Integrations, excisions, truncations, and evolutionary implications. J. Bacteriol. 2003, 185, 3596–3605. [Google Scholar] [CrossRef] [Green Version]
- Ogura, Y.; Ooka, T.; Asadulghani; Terajima, J.; Nougayrède, J.P.; Kurokawa, K.; Tashiro, K.; Tobe, T.; Nakayama, K.; Kuhara, S.; et al. Extensive genomic diversity and selective conservation of virulence-determinants in enterohemorrhagic Escherichia coli strains of O157 and non-O157 serotypes. Genome Biol. 2007, 8. [Google Scholar] [CrossRef] [Green Version]
- Steyert, S.R.; Sahl, J.W.; Fraser, C.M.; Teel, L.D.; Scheutz, F.; Rasko, D.A. Comparative genomics and Stx phage characterization of LEE-negative Shiga toxin-producing Escherichia coli. Front. Cell. Infect. Microbiol. 2012, 2, 133. [Google Scholar] [CrossRef] [Green Version]
- Kulasekara, B.R.; Jacobs, M.; Zhou, Y.; Wu, Z.; Sims, E.; Saenphimmachak, C.; Rohmer, L.; Ritchie, J.M.; Radey, M.; McKevitt, M.; et al. Analysis of the genome of the Escherichia coli O157:H7 2006 spinach-associated outbreak isolate indicates candidate genes that may enhance virulence. Infect. Immun. 2009, 77, 3713–3721. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Makino, K.; Ohnishi, M.; Kurokawa, K.; Ishii, K.; Yokoyama, K.; Han, C.G.; Ohtsubo, E.; Nakayama, K.; Murata, T.; et al. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 2001, 8, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Perna, N.T.; Plunkett, G.; Burland, V.; Mau, B.; Glasner, J.D.; Rose, D.J.; Mayhew, G.F.; Evans, P.S.; Gregor, J.; Kirkpatrick, H.A.; et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 2001, 409, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra-Moreno, R.; Jofre, J.; Muniesa, M. The CI repressors of Shiga toxin-converting prophages are involved in coinfection of Escherichia coli strains, which causes a down regulation in the production of Shiga toxin 2. J. Bacteriol. 2008, 190, 4722–4735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra-Moreno, R.; Jofre, J.; Muniesa, M. Insertion site occupancy by Stx2 bacteriophages depends on the locus availability of the host strain chromosome. J. Bacteriol. 2007, 189, 6645–6654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushiro, A.; Sato, K.; Miyamoto, H.; Yamamura, T.; Honda, T. Induction of prophages of enterohemorrhagic Escherichia coli O157:H7 with norfloxacin. J. Bacteriol. 1999, 181, 2257–2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Ali, G.S.; Ouellette, L.M.; Henderson, S.T.; Lacher, D.W.; Riordan, J.T.; Whittam, T.S.; Manning, S.D. Increased adherence and expression of virulence genes in a lineage of Escherichia coli O157:H7 commonly associated with human infections. PLoS ONE 2010, 5, e10167. [Google Scholar] [CrossRef]
- Neupane, M.; Abu-Ali, G.S.; Mitra, A.; Lacher, D.W.; Manning, S.D.; Riordan, J.T. Shiga toxin 2 overexpression in Escherichia coli O157:H7 strains associated with severe human disease. Microb. Pathog. 2011, 51, 466–470. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Laing, C.; Zhang, Z.; Hallewell, J.; You, C.; Ziebell, K.; Johnson, R.P.; Kropinski, A.M.; Thomas, J.E.; Karmali, M.; et al. Lineage and host source are both correlated with levels of Shiga toxin 2 production by Escherichia coli O157:H7 strains. Appl. Environ. Microbiol. 2010, 76, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Besser, T.E.; Shaikh, N.; Holt, N.J.; Tarr, P.I.; Konkel, M.E.; Malik-Kale, P.; Walsh, C.W.; Whittam, T.S.; Bono, J.L. Greater diversity of Shiga toxin-encoding bacteriophage insertion sites among Escherichia coli O157:H7 isolates from cattle than in those from humans. Appl. Environ. Microbiol. 2007, 73, 671–679. [Google Scholar] [CrossRef] [Green Version]
- Manning, S.D.; Motiwala, A.S.; Springman, A.C.; Qi, W.; Lacher, D.W.; Ouellette, L.M.; Mladonicky, J.M.; Somsel, P.; Rudrik, J.T.; Dietrich, S.E.; et al. Variation in virulence among clades of Escherichia coli O157:H7 associated with disease outbreaks. Proc. Natl. Acad. Sci. USA 2008, 105, 4868–4873. [Google Scholar] [CrossRef] [Green Version]
- Clawson, M.L.; Keen, J.E.; Smith, T.P.L.; Durso, L.M.; McDaneld, T.G.; Mandrell, R.E.; Davis, M.A.; Bono, J.L. Phylogenetic classification of Escherichia coli O157:H7 strains of human and bovine origin using a novel set of nucleotide polymorphisms. Genome Biol. 2009, 10, R56. [Google Scholar] [CrossRef] [Green Version]
- Bono, J.L.; Smith, T.P.; Keen, J.E.; Harhay, G.P.; McDaneld, T.G.; Mandrell, R.E.; Jung, W.K.; Besser, T.E.; Gerner-Smidt, P.; Bielaszewska, M.; et al. Phylogeny of Shiga toxin-producing Escherichia coli O157 isolated from cattle and clinically ill humans. Mol. Biol. Evol. 2012, 29, 2047–2062. [Google Scholar] [CrossRef] [Green Version]
- Skinner, L.M.; Jackson, M.P. Investigation of ribosome binding by the Shiga toxin A1 subunit, using competition and site-directed mutagenesis. J. Bacteriol. 1997, 179, 1368–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohnishi, M.; Kurokawa, K.; Hayashi, T. Diversification of Escherichia coli genomes: Are bacteriophages the major contributors? Trends Microbiol. 2001, 9, 481–485. [Google Scholar] [CrossRef]
- Shaaban, S.; Cowley, L.A.; McAteer, S.P.; Jenkins, C.; Dallman, T.J.; Bono, J.L.; Gally, D.L. Evolution of a zoonotic pathogen: Investigating prophage diversity in enterohaemorrhagic Escherichia coli O157 by long-read sequencing. Microb. Genom. 2016, 2, e000096. [Google Scholar] [CrossRef]
- Ogura, Y.; Seto, K.; Morimoto, Y.; Nakamura, K.; Sato, M.P.; Gotoh, Y.; Itoh, T.; Toyoda, A.; Ohnishi, M.; Hayashi, T. Genomic characterization of β-glucuronidase–positive Escherichia coli O157:H7 producing Stx2a. Emerg. Infect. Dis. 2018, 24, 2219–2227. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rubio, L.; Haarmann, N.; Schwidder, M.; Muniesa, M.; Schmidt, H. Bacteriophages of Shiga toxin-producing Escherichia coli and their contribution to pathogenicity. Pathogens 2021, 10, 404. [Google Scholar] [CrossRef] [PubMed]
- Asadulghani, M.; Ogura, Y.; Ooka, T.; Itoh, T.; Sawaguchi, A.; Iguchi, A.; Nakayama, K.; Hayashi, T. The defective prophage pool of Escherichia coli O157: Prophage-prophage interactions potentiate horizontal transfer of virulence determinants. PLoS Pathog. 2009, 5, e1000408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogura, Y.; Ooka, T.; Iguchi, A.; Toh, H.; Asadulghani, M.; Oshima, K.; Kodama, T.; Abe, H.; Nakayama, K.; Kurokawa, K.; et al. Comparative genomics reveal the mechanism of the parallel evolution of O157 and non-O157 enterohemorrhagic Escherichia coli. Proc. Natl. Acad. Sci. USA 2009, 106, 17939–17944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, P.L.; Neely, M.N.; Zhang, X.; Acheson, D.W.K.; Waldor, M.K.; Friedman, D.I. Role for a phage promoter in Shiga toxin 2 expression from a pathogenic Escherichia coli strain. J. Bacteriol. 2001, 183, 2081–2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, P.L.; Acheson, D.W.K.; Waldor, M.K. Isogenic lysogens of diverse Shiga toxin 2-encoding bacteriophages produce markedly different amounts of Shiga toxin. Infect. Immun. 1999, 67, 6710–6714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, S.; Rusconi, B.; Sanjar, F.; Goswami, K.; Xiaoli, L.; Eppinger, M.; Dudley, E.G. Escherichia coli O157: H7 strains harbor at least three distinct sequence types of Shiga toxin 2a-converting phages. BMC Genom. 2015, 16, 733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, S.D.; Madera, R.T.; Schneider, W.; Dietrich, S.E.; Khalife, W.; Brown, W.; Whittam, T.S.; Somsel, P.; Rudrik, J.T. Surveillance for Shiga toxin-producing Escherichia coli, Michigan, 2001–2005. Emerg. Infect. Dis. 2007, 13, 318–321. [Google Scholar] [CrossRef]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.; Costantino, N.; Court, D.L. A set of recombineering plasmids for gram-negative bacteria. Gene 2006, 379, 109–115. [Google Scholar] [CrossRef] [PubMed]
- De Sablet, T.; Bertin, Y.; Vareille, M.; Girardeau, J.P.; Garrivier, A.; Gobert, A.P.; Martin, C. Differential expression of stx2 variants in Shiga toxin-producing Escherichia coli belonging to seropathotypes A and C. Microbiology 2008, 154, 176–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Ali, G.S.; Ouellette, L.M.; Henderson, S.T.; Whittam, T.S.; Manning, S.D. Differences in adherence and virulence gene expression between two outbreak strains of enterohaemorrhagic Escherichia coli O157:H7. Microbiology 2010, 156, 408–419. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]





| yehV | argW | wrbA | sbcB | ||||||
|---|---|---|---|---|---|---|---|---|---|
| stx Profile | Profile 1 | Stx1a Phage | Non-Stx1a Phage | Stx2a/2c Phage | Non-Stx2a/2c Phage | Stx2a/2c Phage | Non-Stx2a/2c Phage | Stx2a/2c Phage | Non-Stx2a/2c Phage |
| 1a (n = 2) | OIOI | 2 | 0 | - | - | 0 | 2 | - | - |
| 1a, 2a (n = 11) | OIOO | 8 | 0 | 0 | 0 | 8 | 0 | 0 | 8 |
| OIOO | 1 | 0 | - | - | 0 | 1 | 1 | 0 | |
| OIOO * | 2 | 0 | - | - | 0 | 2 | 0 | 2 | |
| 1a, 2a (n = 15) | OOOI ** | 0 | 1 | 1 | 0 | 1 | 0 | - | - |
| OOOI | 11 | 0 | 0 | 11 | 11 | 0 | - | - | |
| OOOI | 3 | 0 | 3 | 0 | 0 | 3 | - | - | |
| 1a, 2c (n = 2) | OOIO | 1 | 0 | 1 | 0 | - | - | 0 | 1 |
| OOIO | 1 | 0 | 0 | 1 | - | - | 1 | 0 | |
| 2a (n = 3) | OIOI | 0 | 3 | - | - | 3 | 0 | - | - |
| 2a (n = 31) | OOII * | 0 | 5 | 0 | 5 | - | - | - | - |
| OOII | 0 | 26 | 26 | 0 | - | - | - | - | |
| 2a (n = 4) | OOIO | 0 | 4 | 4 | 0 | - | - | 0 | 4 |
| 2a, 2c (n = 1) | OIOO * | 0 | 1 | - | - | 0 | 1 | 0 | 1 |
| 2a, 2c (n = 37) | OOIO * | 0 | 2 | 0 | 2 | - | - | 0 | 2 |
| OOIO * | 0 | 6 | 6 | 0 | - | - | 0 | 6 | |
| OOIO | 0 | 26 | 26 | 0 | - | - | 26 | 0 | |
| OOIO * | 0 | 3 | 0 | 3 | - | - | 3 | 0 | |
| 2c (n = 1) | IOOO | - | - | 0 | 1 | 0 | 1 | 1 | 0 |
| 2c (n = 17) | OIIO * | 0 | 9 | - | - | - | - | 0 | 9 |
| OIIO | 0 | 8 | - | - | - | - | 8 | 0 | |
| 2c (n = 1) | OOIO | 0 | 1 | 0 | 1 | - | - | 1 | 0 |
| Total | (n = 125) | 29 | 95 | 67 | 24 | 23 | 10 | 41 | 33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henderson, S.T.; Singh, P.; Knupp, D.; Lacher, D.W.; Abu-Ali, G.S.; Rudrik, J.T.; Manning, S.D. Variability in the Occupancy of Escherichia coli O157 Integration Sites by Shiga Toxin-Encoding Prophages. Toxins 2021, 13, 433. https://doi.org/10.3390/toxins13070433
Henderson ST, Singh P, Knupp D, Lacher DW, Abu-Ali GS, Rudrik JT, Manning SD. Variability in the Occupancy of Escherichia coli O157 Integration Sites by Shiga Toxin-Encoding Prophages. Toxins. 2021; 13(7):433. https://doi.org/10.3390/toxins13070433
Chicago/Turabian StyleHenderson, Scott T., Pallavi Singh, David Knupp, David W. Lacher, Galeb S. Abu-Ali, James T. Rudrik, and Shannon D. Manning. 2021. "Variability in the Occupancy of Escherichia coli O157 Integration Sites by Shiga Toxin-Encoding Prophages" Toxins 13, no. 7: 433. https://doi.org/10.3390/toxins13070433
APA StyleHenderson, S. T., Singh, P., Knupp, D., Lacher, D. W., Abu-Ali, G. S., Rudrik, J. T., & Manning, S. D. (2021). Variability in the Occupancy of Escherichia coli O157 Integration Sites by Shiga Toxin-Encoding Prophages. Toxins, 13(7), 433. https://doi.org/10.3390/toxins13070433