Stability and Safety of Inhibitor Cystine Knot Peptide, GTx1-15, from the Tarantula Spider Grammostola rosea

Abstract
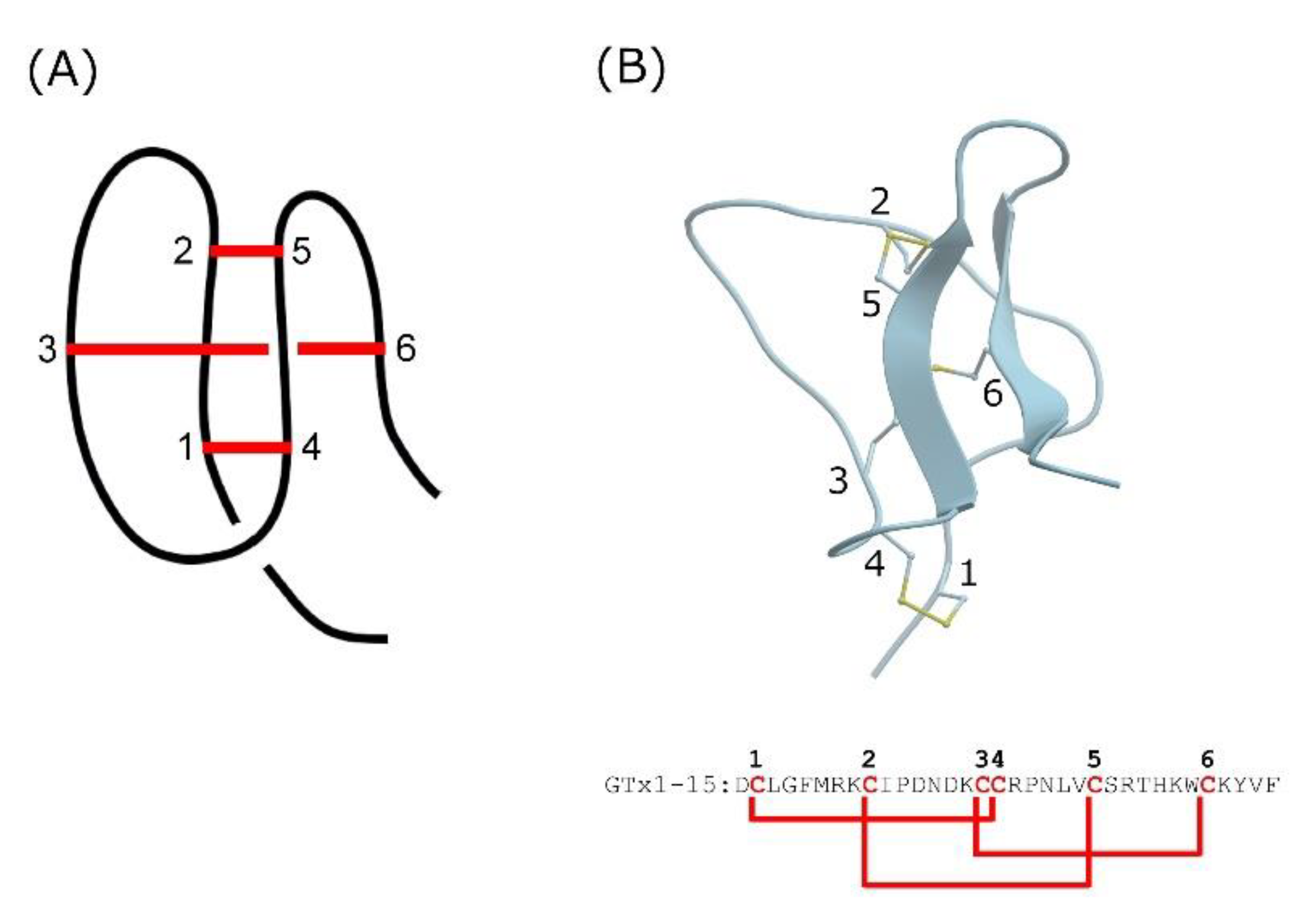
:1. Introduction
2. Results
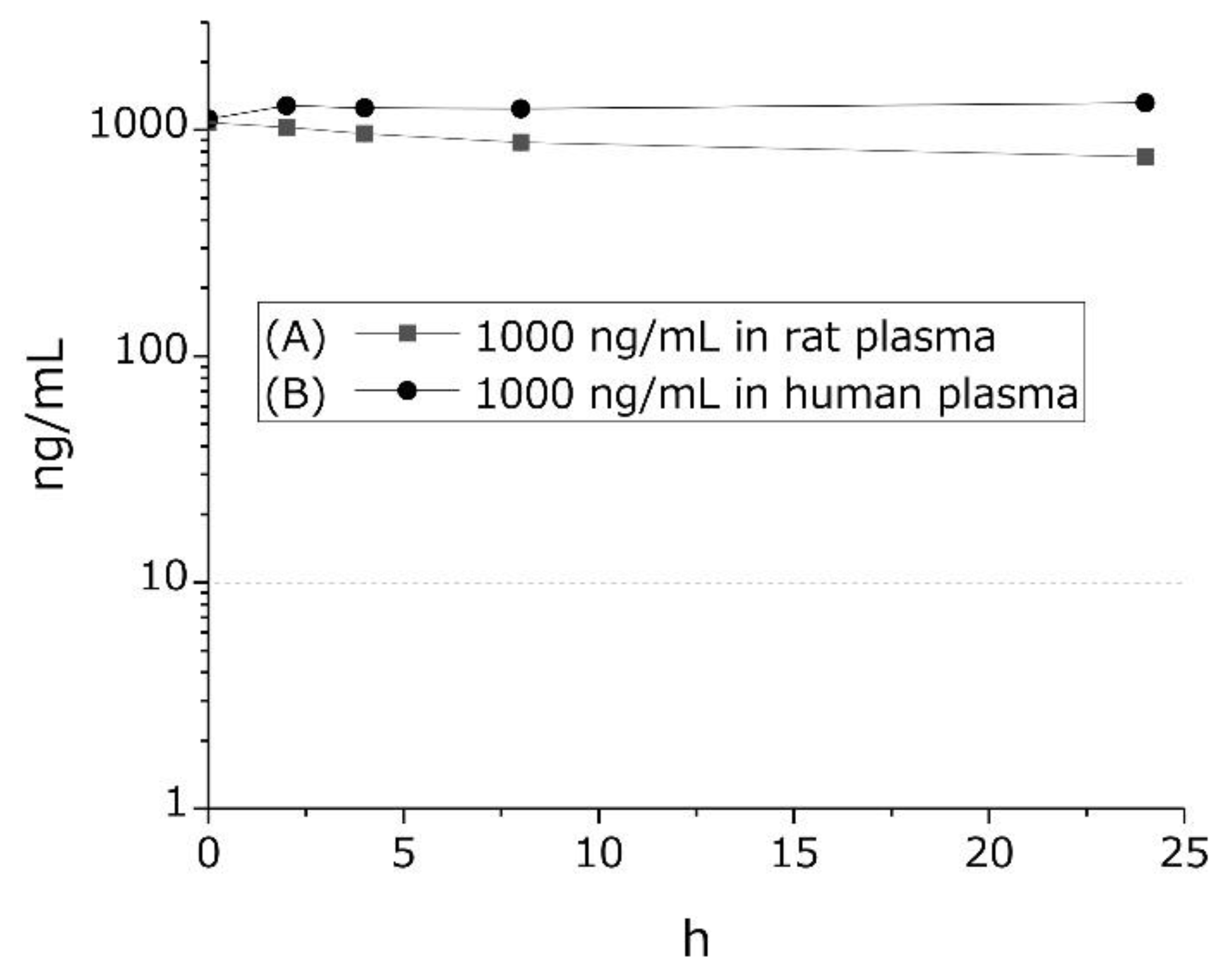
2.1. GTx1-15 Is Not Degraded in Plasma
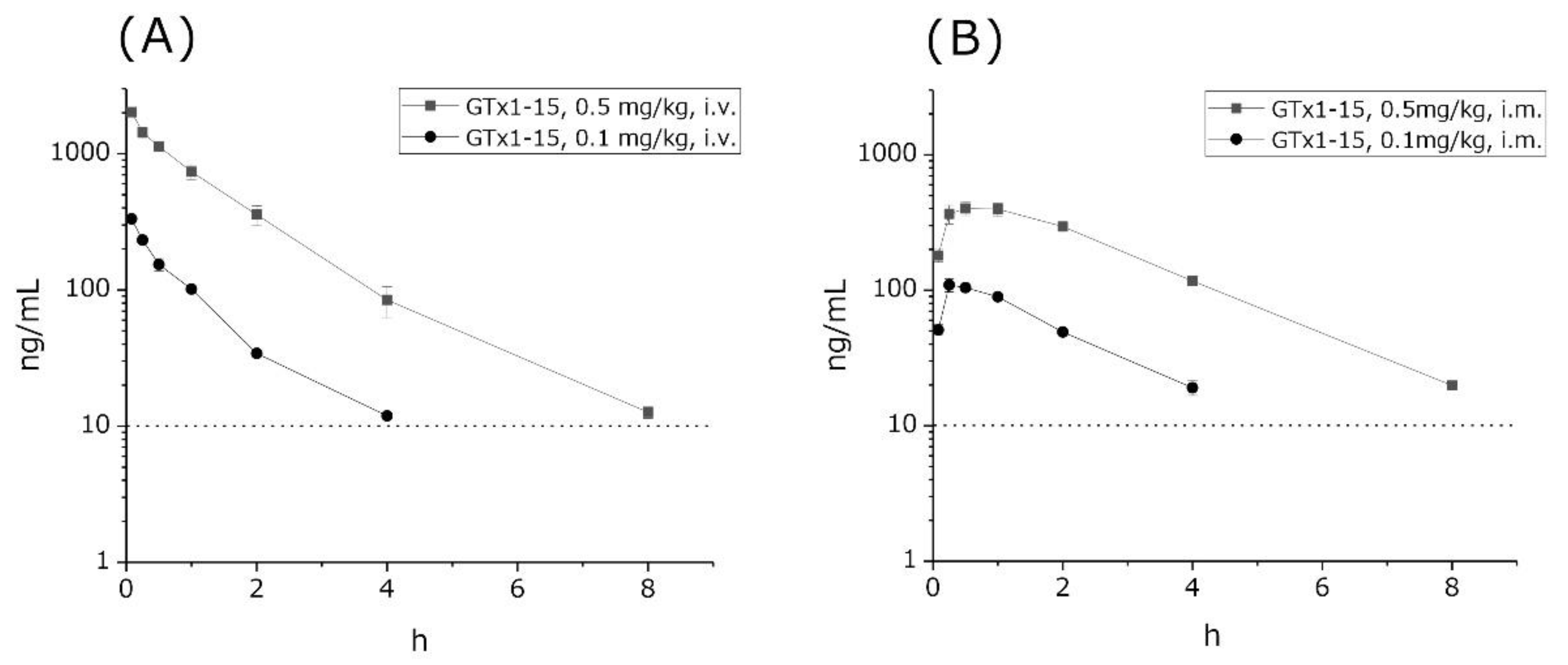
2.2. GTx1-15 Is Stable in Circulation
2.3. GTx1-15 Is Stable in High-Temperature Environments
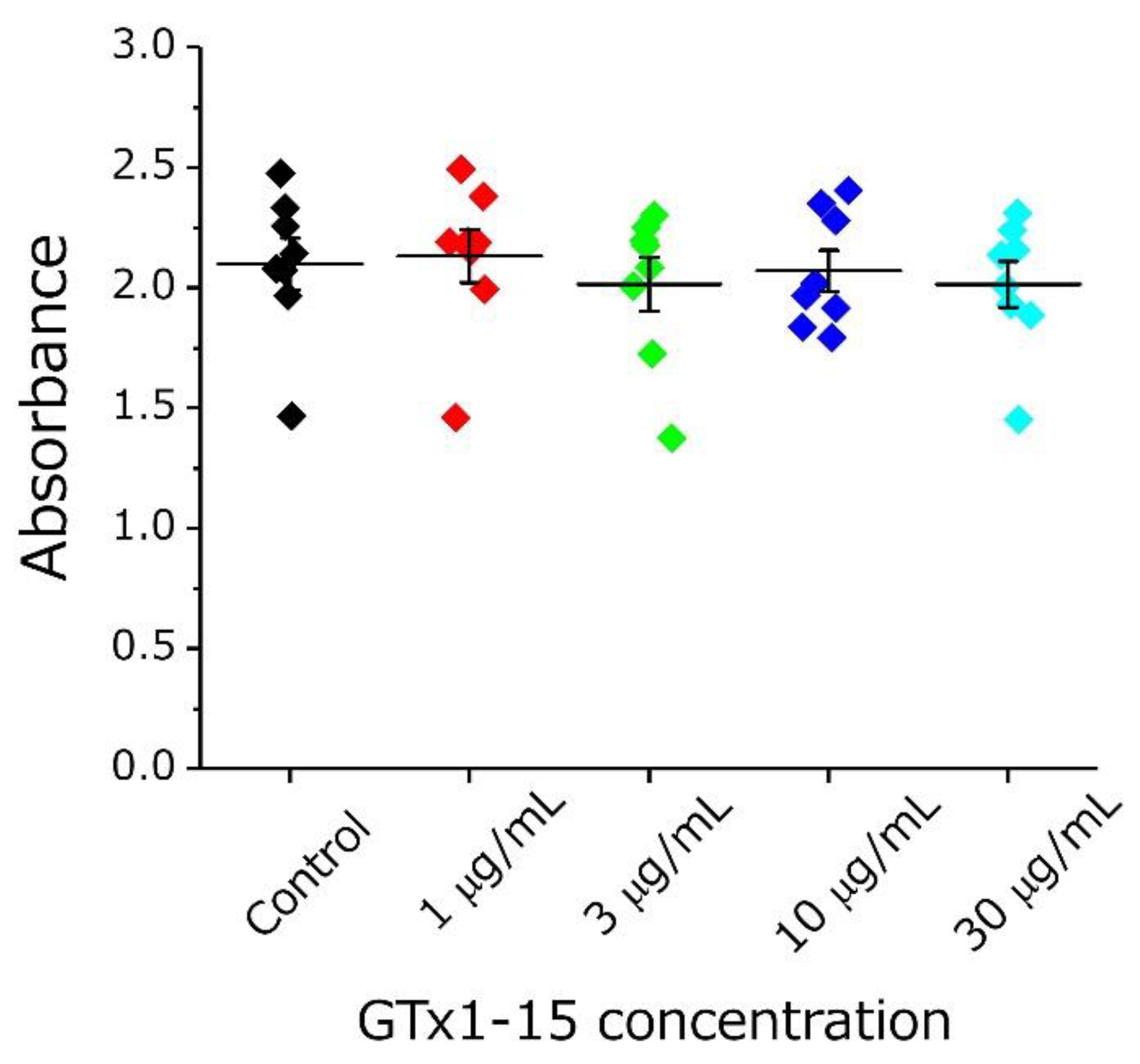
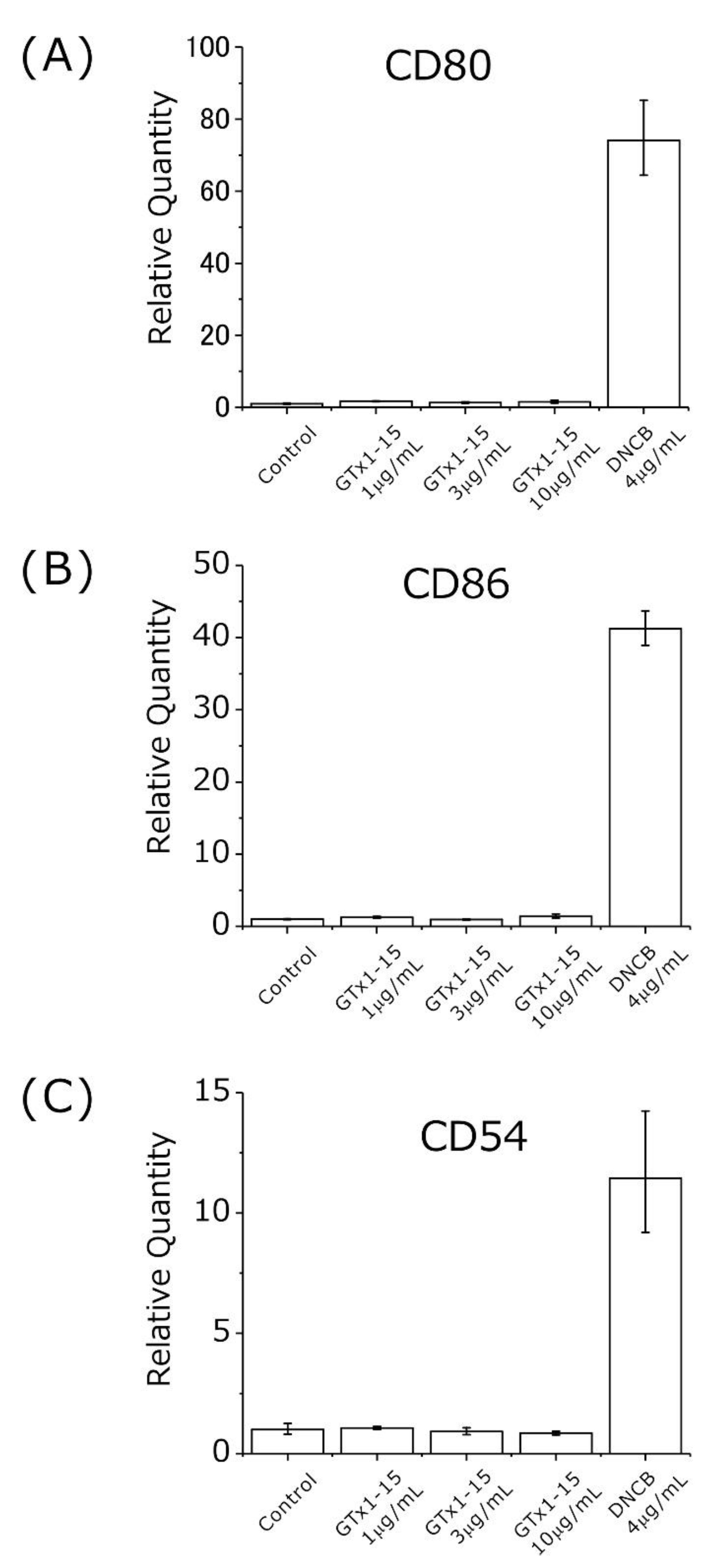
2.4. GTx1-15 Shows No Cytotoxicity or Antigenicity
3. Discussion
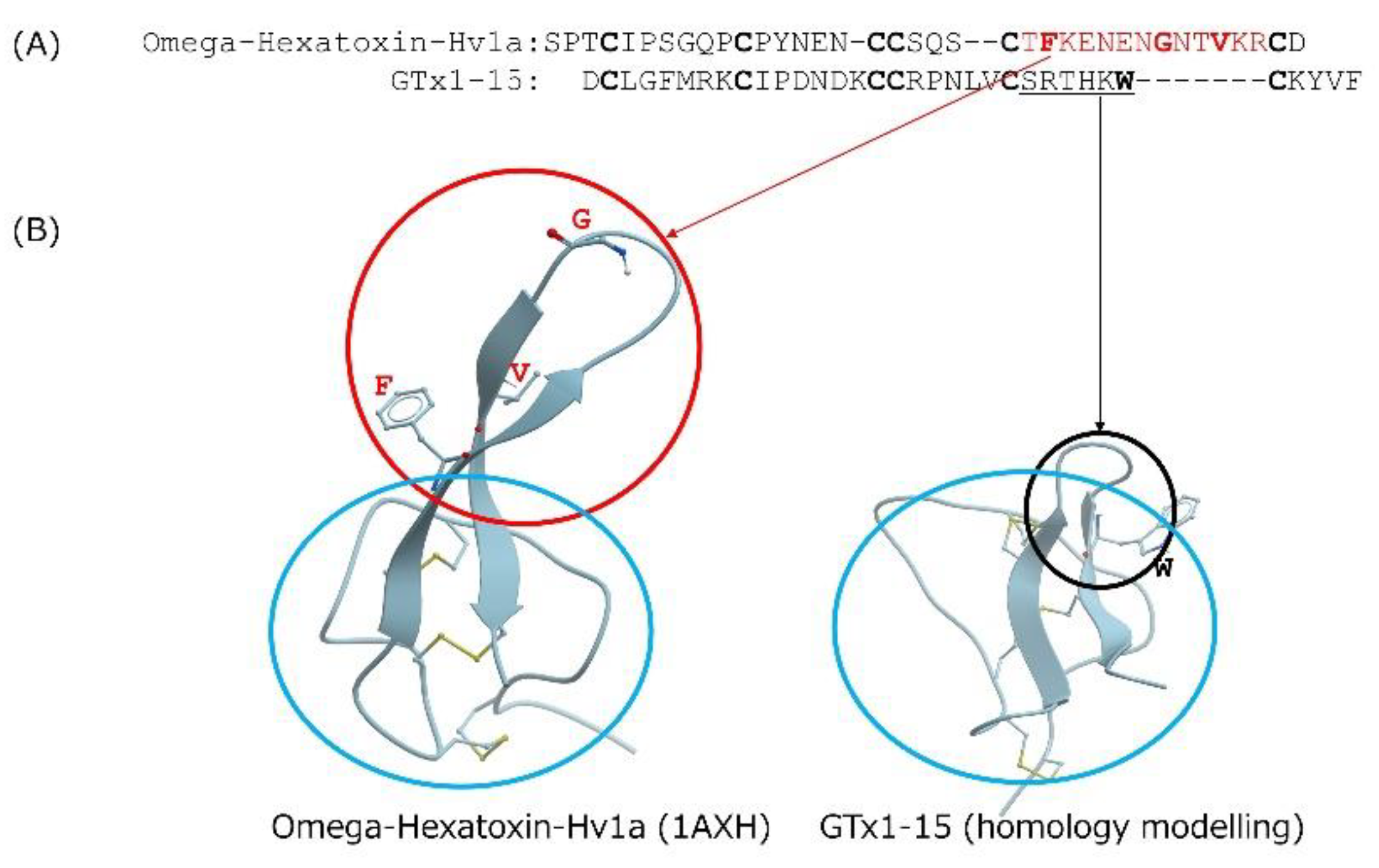
3.1. The GTx1-15 Molecule Shows High Stability
3.2. GTx1-15 Is Safe with No Cytotoxicity or Antigenicity
3.3. GTx1-15 Is a Suitable Scaffold for Peptide Library Construction
4. Conclusions
5. Materials and Methods
5.1. Peptides and Chemicals
5.2. Stability Assays in Animal Plasma In Vitro
5.3. Concentration in Circulation after Intravenous, Intramuscular, and Peroral Administration
5.4. LC-MS/MS
5.5. Thermal Stability
5.6. Protein Thermal Shift Assay
5.7. Cytotoxicity Assay
5.8. Antigenicity Assay
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Annette, C.D.; Paul, A.I.; Terrence, F.B.; Urs, A.M. Introduction to the Theme “Ion Channels and Neuropharmacology: From the Past to the Future”. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 1–6. [Google Scholar] [CrossRef]
- Lazniewska, J.; Weiss, N. Glycosylation of voltage-gated calcium channels in health and disease. Biochim. Biophys. Acta 2017, 1859, 662–668. [Google Scholar] [CrossRef]
- Todorovic, S.M.; Jevtovic-Todorovic, V. Targeting of Cav3.2 T-type calcium channels in peripheral sensory neurons for the treatment of painful diabetic neuropathy. Pflügers Arch. 2014, 466, 701–706. [Google Scholar] [CrossRef]
- Lang, F.; Stournaras, C. Ion channels in cancer: Future perspectives and clinical potential. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130108. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, A.; Saha, S. T-Type voltage gated calcium channels: A target in breast cancer? Breast Cancer Res. Treat. 2019, 173, 11–21. [Google Scholar] [CrossRef]
- McGivern, J.G. Ziconotide: A review of its pharmacology and use in the treatment of pain. Neuropsychiatr. Dis. Treat. 2007, 3, 69–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raufman, J.P. Bioactive peptides from lizard venoms. Regul. Pept. 1996, 61, 1–18. [Google Scholar] [CrossRef]
- Prashanth, J.R.; Hasaballah, N.; Vetter, I. Pharmacological screening technologies for venom peptide discovery. Neuropharmacology 2017, 127, 4–19. [Google Scholar] [CrossRef]
- Doupnik, C.A. Venom-derived peptides inhibiting Kir channels: Past, present, and future. Neuropharmacology 2017, 127, 161–172. [Google Scholar] [CrossRef]
- Ma, R.; Mahadevappa, R.; Kwok, H.F. Venom-based peptide therapy: Insights into anti-cancer mechanism. Oncotarget 2017, 8, 100908–100930. [Google Scholar] [CrossRef] [Green Version]
- Escoubas, P.; Sollod, B.; King, G.F. Venom landscapes: Mining the complexity of spider venoms via a combined cDNA and mass spectrometric approach. Toxicon 2006, 47, 650–663. [Google Scholar] [CrossRef]
- Pennington, M.W.; Czerwinski, A.; Norton, R.S. Peptide therapeutics from venom: Current status and potential. Bioorg. Med. Chem. 2018, 26, 2738–2758. [Google Scholar] [CrossRef]
- Saez, N.J.; Herzig, V. Versatile spider venom peptides and their medical and agricultural applications. Toxicon 2019, 158, 109–126. [Google Scholar] [CrossRef]
- Herzig, V.; Cristofori-Armstrong, B.; Israel, M.R.; Nixon, S.A.; Vetter, I.; King, G.F. Animal toxins—Nature’s evolutionary-refined toolkit for basic research and drug discovery. Biochem. Pharmacol. 2020, 181, 114096. [Google Scholar] [CrossRef] [PubMed]
- Colgrave, M.L.; Craik, D.J. Thermal, Chemical, and Enzymatic Stability of the Cyclotide Kalata B1: The Importance of the Cyclic Cystine Knot. Biochemistry 2004, 43, 5965–5975. [Google Scholar] [CrossRef]
- Herzig, V.; King, G. The Cystine Knot Is Responsible for the Exceptional Stability of the Insecticidal Spider Toxin ω-Hexatoxin-Hv1a. Toxins 2015, 7, 4366–4380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postic, G.; Gracy, J.; Périn, C.; Chiche, L.; Gelly, J.-C. KNOTTIN: The database of inhibitor cystine knot scaffold after 10 years, toward a systematic structure modeling. Nucleic Acids Res. 2018, 46, D454–D458. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, N.; Wu, B.; Ligutti, J.; Cheneval, O.; Agwa, A.J.; Benfield, A.H.; Biswas, K.; Craik, D.J.; Miranda, L.P.; Henriques, S.T.; et al. Peptide-Membrane Interactions Affect the Inhibitory Potency and Selectivity of Spider Toxins ProTx-II and GpTx-1. ACS Chem. Biol. 2019, 14, 118–130. [Google Scholar] [CrossRef]
- Mobli, M.; Undheim, E.A.B.; Rash, L.D. Modulation of Ion Channels by Cysteine-Rich Peptides: From Sequence to Structure. Adv. Pharmacol. 2017, 79, 199–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, S.; Kimura, T.; Kubo, T. Characterization of voltage-dependent calcium channel blocking peptides from the venom of the tarantula Grammostola rosea. Toxicon 2011, 58, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Meir, A.; Cheriki, R.S.; Kolb, E.; Langut, Y.; Bajayo, N. Novel Peptides Isolated from Spider Venom, and Uses Thereof PCT Patent Application Publication WO/2011/033358, 24 March 2011.
- Kikuchi, K.; Sugiura, M.; Kimura, T. High Proteolytic Resistance of Spider-Derived Inhibitor Cystine Knots. Int. J. Pept. 2015, 2015, 537508. [Google Scholar] [CrossRef] [Green Version]
- Bassotti, G.; Usai-Satta, P.; Bellini, M. Linaclotide for the treatment of chronic constipation. Expert Opin. Pharmacother. 2018, 19, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, H.; Ashikaga, T.; Miyazawa, M.; Kosaka, N.; Ito, Y.; Yoneyama, K.; Sono, S.; Itagaki, H.; Toyoda, H.; Suzuki, H. The relationship between CD86/CD54 expression and THP-1 cell viability in an in vitro skin sensitization test—Human cell line activation test (h-CLAT). Cell Biol. Toxicol. 2009, 25, 109–126. [Google Scholar] [CrossRef]
- Bocchietto, E.; Paolucci, C.; Breda, D.; Sabbioni, E.; Burastero, S.E. Human monocytoid THP-1 cell line versus monocyte-derived human immature dendritic cells as in vitro models for predicting the sensitising potential of chemicals. Int. J. Immunopathol. Pharmacol. 2007, 20, 259–265. [Google Scholar] [CrossRef] [Green Version]
- Agwa, A.J.; Tran, P.; Mueller, A.; Tran, H.N.T.; Deuis, J.R.; Israel, M.R.; McMahon, K.L.; Craik, D.J.; Vetter, I.; Schroeder, C.I. Manipulation of a spider peptide toxin alters its affinity for lipid bilayers and potency and selectivity for voltage-gated sodium channel subtype 1.7. J. Biol. Chem. 2020, 295, 5067–5080. [Google Scholar] [CrossRef]
- Werle, M.; Schmitz, T.; Huang, H.L.; Wentzel, A.; Kolmar, H.; Bernkop-Schnurch, A. The potential of cystine-knot microproteins as novel pharmacophoric scaffolds in oral peptide drug delivery. J. Drug Target. 2006, 14, 137–146. [Google Scholar] [CrossRef]
- Newcomb, R.; Abbruscato, T.J.; Singh, T.; Nadasdi, L.; Davis, T.P.; Miljanich, G. Bioavailability of Ziconotide in brain: Influx from blood, stability, and diffusion. Peptides 2000, 21, 491–501. [Google Scholar] [CrossRef]
- Wang, D.; Starr, R.; Chang, W.C.; Aguilar, B.; Alizadeh, D.; Wright, S.L.; Yang, X.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Falcao, C.B.; Radis-Baptista, G. Crotamine and crotalicidin, membrane active peptides from Crotalus durissus terrificus rattlesnake venom, and their structurally-minimized fragments for applications in medicine and biotechnology. Peptides 2020, 126, 170234. [Google Scholar] [CrossRef] [PubMed]
- Sansom, D.M.; Manzotti, C.N.; Zheng, Y. What’s the difference between CD80 and CD86? Trends Immunol. 2003, 24, 314–319. [Google Scholar] [CrossRef]
- Stucki, A.; Rivier, A.S.; Gikic, M.; Monai, N.; Schapira, M.; Spertini, O. Endothelial cell activation by myeloblasts: Molecular mechanisms of leukostasis and leukemic cell dissemination. Blood 2001, 97, 2121–2129. [Google Scholar] [CrossRef] [PubMed]
- Corti, D.; Galbiati, V.; Gatti, N.; Marinovich, M.; Galli, C.L.; Corsini, E. Optimization of the THP-1 activation assay to detect pharmaceuticals with potential to cause immune mediated drug reactions. Toxicol. In Vitro 2015, 29, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T. Screening Techniques Using the Periplasmic Expression of Peptide Libraries and Target Molecules. J. Bioanal. Biomed. 2017, 9. [Google Scholar] [CrossRef]
- Kikuchi, K.; Sugiura, M.; Nishizawa-Harada, C.; Kimura, T. The application of the Escherichia coli giant spheroplast for drug screening with automated planar patch clamp system. Biotechnol. Rep. 2015, 7, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Turchetto, J.; Sequeira, A.F.; Ramond, L.; Peysson, F.; Brás, J.L.A.; Saez, N.J.; Duhoo, Y.; Blémont, M.; Guerreiro, C.I.P.D.; Quinton, L.; et al. High-throughput expression of animal venom toxins in Escherichia coli to generate a large library of oxidized disulphide-reticulated peptides for drug discovery. Microb. Cell Factories 2017, 16, 6. [Google Scholar] [CrossRef] [Green Version]
- Sequeira, A.F.; Turchetto, J.; Saez, N.J.; Peysson, F.; Ramond, L.; Duhoo, Y.; Blémont, M.; Fernandes, V.O.; Gama, L.T.; Ferreira, L.M.A.; et al. Gene design, fusion technology and TEV cleavage conditions influence the purification of oxidized disulphide-rich venom peptides in Escherichia coli. Microb. Cell Factories 2017, 16, 4. [Google Scholar] [CrossRef] [Green Version]
- Saez, N.J.; Cristofori-Armstrong, B.; Anangi, R.; King, G.F. A Strategy for Production of Correctly Folded Disulfide-Rich Peptides in the Periplasm of E. coli. Methods Mol. Biol. 2017, 1586, 155–180. [Google Scholar] [CrossRef]
- Klint, J.K.; Senff, S.; Saez, N.J.; Seshadri, R.; Lau, H.Y.; Bende, N.S.; Undheim, E.A.; Rash, L.D.; Mobli, M.; King, G.F. Production of recombinant disulfide-rich venom peptides for structural and functional analysis via expression in the periplasm of E. coli. PLoS ONE 2013, 8, e63865. [Google Scholar] [CrossRef] [Green Version]
- Craik, D.J.; Daly, N.L.; Waine, C. The cystine knot motif in toxins and implications for drug design. Toxicon 2001, 39, 43–60. [Google Scholar] [CrossRef]
- Heitz, A.; Avrutina, O.; Le-Nguyen, D.; Diederichsen, U.; Hernandez, J.F.; Gracy, J.; Kolmar, H.; Chiche, L. Knottin cyclization: Impact on structure and dynamics. BMC Struct. Biol. 2008, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Kolmar, H. Biological diversity and therapeutic potential of natural and engineered cystine knot miniproteins. Curr. Opin. Pharmacol. 2009, 9, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Reinwarth, M.; Nasu, D.; Kolmar, H.; Avrutina, O. Chemical synthesis, backbone cyclization and oxidative folding of cystine-knot peptides: Promising scaffolds for applications in drug design. Molecules 2012, 17, 12533–12552. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T. Screening and Fast Identification of ICK Peptides Which Affect Kv2.1 Activity Using Periplasmic Peptide Display Technique PERISS; National Institute of Advanced Industrial Science and Technology (AIST): Tsukuba, Japan, 2021; manuscript in preparation. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ICK Peptide | Tm B (°C) |
|---|---|
| GTx1-15 | 96.30 ± 0.03 |
| ProTxI | 93.23 ± 0.12 |
| ProTxII | 93.95 ± 0.12 |
| GsMTx4 | 95.26 ± 0.10 |
| Gene | Sequence | Amplicon Size (bp) | |
|---|---|---|---|
| CD80 | Forward Reverse | CCTACTGCTTTGCCCCAAGA AAGGGCAAGGTGGGGTAATC | 188 |
| CD86 | Forward Reverse | ACGCGGCTTTTATCTTCACC TCTTCCCTCTCCATTGTGTTGG | 200 |
| CD54 | Forward Reverse | TTGAGGGCACCTACCTCTGT GATCTTCCGCTGGCGGTTAT | 176 |
| GAPDH | Forward Reverse | CCATGGAGAAGGCTGGGG CAAAGTTGTCATGGATGACC | 195 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimura, T. Stability and Safety of Inhibitor Cystine Knot Peptide, GTx1-15, from the Tarantula Spider Grammostola rosea. Toxins 2021, 13, 621. https://doi.org/10.3390/toxins13090621
Kimura T. Stability and Safety of Inhibitor Cystine Knot Peptide, GTx1-15, from the Tarantula Spider Grammostola rosea. Toxins. 2021; 13(9):621. https://doi.org/10.3390/toxins13090621
Chicago/Turabian StyleKimura, Tadashi. 2021. "Stability and Safety of Inhibitor Cystine Knot Peptide, GTx1-15, from the Tarantula Spider Grammostola rosea" Toxins 13, no. 9: 621. https://doi.org/10.3390/toxins13090621
APA StyleKimura, T. (2021). Stability and Safety of Inhibitor Cystine Knot Peptide, GTx1-15, from the Tarantula Spider Grammostola rosea. Toxins, 13(9), 621. https://doi.org/10.3390/toxins13090621