A Combined Transcriptomics and Proteomics Approach Reveals the Differences in the Predatory and Defensive Venoms of the Molluscivorous Cone Snail Cylinder ammiralis (Caenogastropoda: Conidae)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Composition of C. ammiralis Venom
2.1.1. Venom Gland Transcriptome
2.1.2. Mass Spectrometry Analysis of Predatory and Defensive Venoms
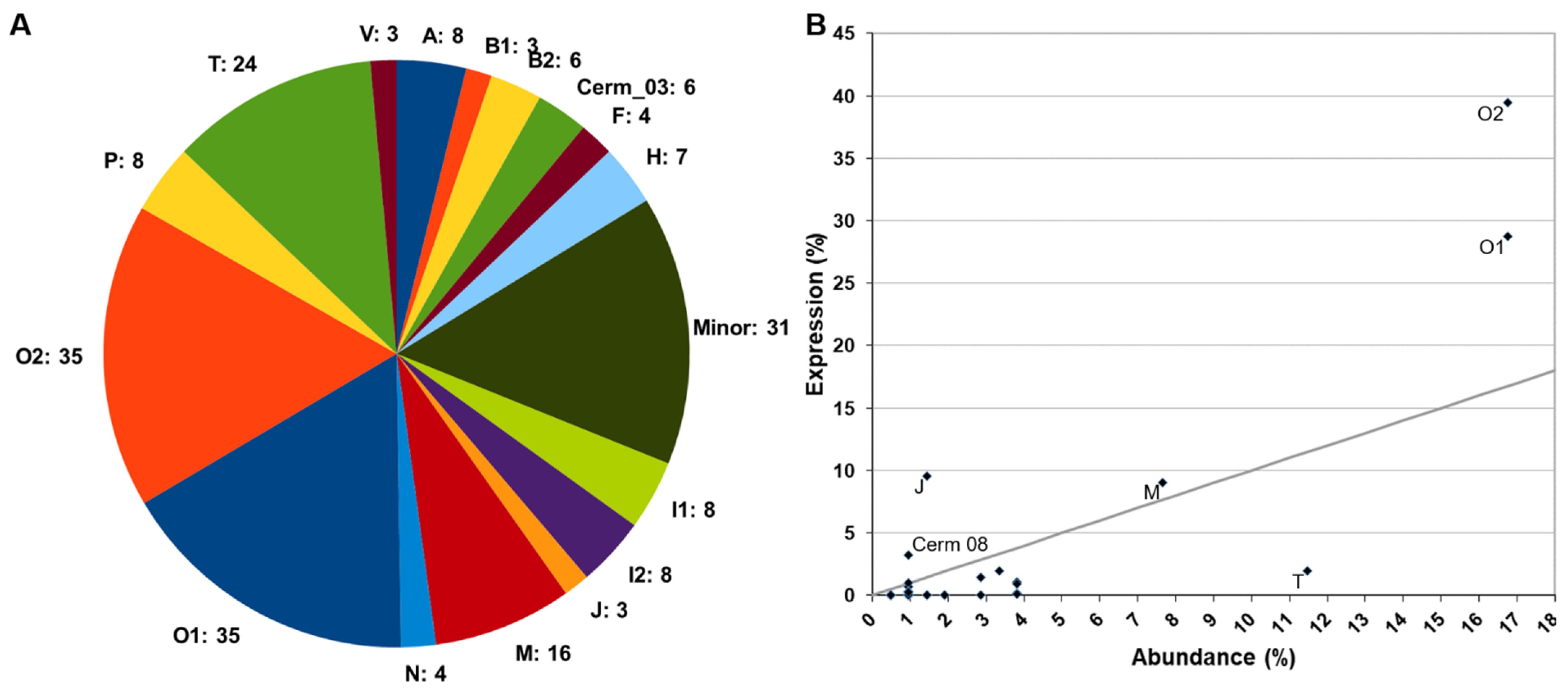
2.1.3. Transcriptome versus Proteomes
2.2. Differences in Venom Composition across Diets
3. Discussion
3.1. Transcriptomics and Proteomics, Two Complementary Approaches to Determine Venom Composition
3.2. Predatory- versus Defensive-Evoked Venoms
3.3. Comparative Analyses of Venom Composition across Diets
4. Conclusions
5. Materials and Methods
5.1. Taxon Sampling
5.2. RNA Extraction and Sequencing
5.3. Transcriptome Assembly and Transcript Annotation
5.4. Venom Milking
5.5. LC–MS and Proteomic Analysis
5.6. Bioinformatic Integration of Proteomic and Transcriptomic Data
5.7. Analysis of Venom Composition across Diets
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Modica, M.V.; Sunagar, K.; Holford, M.; Dutertre, S. Diversity and Evolution of Animal Venoms: Neglected Targets, Ecological Interactions, Future Perspectives. Front. Ecol. Evol. 2020, 8, 65. [Google Scholar] [CrossRef] [Green Version]
- Holford, M.; Daly, M.; King, G.F.; Norton, R.S. Venoms to the rescue. Science 2018, 361, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Suryamohan, K.; Krishnankutty, S.P.; Guillory, J.; Jevit, M.; Schröder, M.S.; Wu, M.; Kuriakose, B.; Mathew, O.K.; Perumal, R.C.; Koludarov, I.; et al. The Indian cobra reference genome and transcriptome enables comprehensive identification of venom toxins. Nat. Genet. 2020, 52, 106–117. [Google Scholar] [CrossRef] [Green Version]
- Pardos-Blas, J.R.; Irisarri, I.; Abalde, S.; Afonso, C.M.L.; Tenorio, M.J.; Zardoya, R. The genome of the venomous snail Lautoconus ventricosus sheds light on the origin of conotoxin diversity. Gigascience 2021, 10, giab037. [Google Scholar] [CrossRef] [PubMed]
- Sanggaard, K.W.; Bechsgaard, J.S.; Fang, X.; Duan, J.; Dyrlund, T.F.; Gupta, V.; Jiang, X.; Cheng, L.; Fan, D.; Feng, Y. Spider genomes provide insight into composition and evolution of venom and silk. Nat. Commun. 2014, 5, 1–12. [Google Scholar]
- Dutertre, S.; Jin, A.-H.; Vetter, I.; Hamilton, B.; Sunagar, K.; Lavergne, V.; Dutertre, V.; Fry, B.G.; Antunes, A.; Venter, D.J. Evolution of separate predation-and defence-evoked venoms in carnivorous cone snails. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- King, G. Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics; Royal Society of Chemistry: London, UK, 2015; ISBN 1849736634. [Google Scholar]
- Wilson, D.; Daly, N.L. Venomics: A mini-review. High-Throughput 2018, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Nagalakshmi, U.; Waern, K.; Snyder, M. RNA-Seq: A method for comprehensive transcriptome analysis. Curr. Protoc. Mol. Biol. 2010, 89, 4–11. [Google Scholar] [CrossRef]
- Barua, A.; Mikheyev, A.S. An ancient, conserved gene regulatory network led to the rise of oral venom systems. Proc. Natl. Acad. Sci. USA 2021, 118, e2021311118. [Google Scholar] [CrossRef]
- Abalde, S.; Tenorio, M.J.; Afonso, C.M.L.; Zardoya, R. Comparative transcriptomics of the venoms of continental and insular radiations of West African cones. Proc. R. Soc. B 2020, 287, 20200794. [Google Scholar] [CrossRef]
- Shyu, A.B.; Wilkinson, M.F.; Van Hoof, A. Messenger RNA regulation: To translate or to degrade. EMBO J. 2008, 27, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Guo, S.; Gao, J.; Luo, L.; Liao, X.; Li, M.; Su, H.; Huang, Z.; Xu, J.; Pei, J. Kinetic analysis of effects of temperature and time on the regulation of venom expression in Bungarus multicinctus. Sci. Rep. 2020, 10, 1–11. [Google Scholar]
- Hofmann, E.P.; Rautsaw, R.M.; Strickland, J.L.; Holding, M.L.; Hogan, M.P.; Mason, A.J.; Rokyta, D.R.; Parkinson, C.L. Comparative venom-gland transcriptomics and venom proteomics of four Sidewinder Rattlesnake (Crotalus cerastes) lineages reveal little differential expression despite individual variation. Sci. Rep. 2018, 8, 1–15. [Google Scholar]
- Abalde, S.; Tenorio, M.J.; Afonso, C.M.L.; Zardoya, R. Conotoxin Diversity in Chelyconus ermineus (Born, 1778) and the Convergent Origin of Piscivory in the Atlantic and Indo-Pacific Cones. Genome Biol. Evol. 2018, 10, 2643–2662. [Google Scholar] [CrossRef] [Green Version]
- Jin, A.-H.; Dutertre, S.; Dutt, M.; Lavergne, V.; Jones, A.; Lewis, R.J.; Alewood, P.F. Transcriptomic-proteomic correlation in the predation-evoked venom of the cone snail, Conus imperialis. Mar. Drugs 2019, 17, 177. [Google Scholar] [CrossRef] [Green Version]
- Monnier, E.; Limpalaër, L.; Robin, A.; Roux, C. A Taxonomic Iconography of Living Conidae; ConchBooks: Harxheim, Germany, 2018; ISBN 9783939767923. [Google Scholar]
- Olivera, B.M.; Seger, J.; Horvath, M.P.; Fedosov, A.E. Prey-capture strategies of fish-hunting cone snails: Behavior, neurobiology and evolution. Brain. Behav. Evol. 2015, 86, 58–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phuong, M.A.; Mahardika, G.N.; Alfaro, M.E. Dietary breadth is positively correlated with venom complexity in cone snails. BMC Genom. 2016, 17, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duda, T.F.; Kohn, A.J.; Palumbi, R.S. Origins of diverse feeding ecologies within Conus, a genus of venomous marine gastropods. Biol. J. Linn. Soc. 2001, 73, 391–409. [Google Scholar] [CrossRef]
- Robinson, S.D.; Norton, R.S. Conotoxin gene superfamilies. Mar. Drugs 2014, 12, 6058–6101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safavi-Hemami, H.; Gajewiak, J.; Karanth, S.; Robinson, S.D.; Ueberheide, B.; Douglass, A.D.; Schlegel, A.; Imperial, J.S.; Watkins, M.; Bandyopadhyay, P.K. Specialized insulin is used for chemical warfare by fish-hunting cone snails. Proc. Natl. Acad. Sci. USA 2015, 112, 1743–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.; Menting, J.G.; Disotuar, M.M.; Smith, N.A.; Delaine, C.A.; Ghabash, G.; Agrawal, R.; Wang, X.; He, X.; Fisher, S.J.; et al. A structurally minimized yet fully active insulin based on cone-snail venom insulin principles. Nat. Struct. Mol. Biol. 2020, 27, 615–624. [Google Scholar] [CrossRef]
- Neves, J.L.B.; Lin, Z.; Imperial, J.S.; Antunes, A.; Vasconcelos, V.; Olivera, B.M.; Schmidt, E.W. Small molecules in the cone snail arsenal. Org. Lett. 2015, 17, 4933–4935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.; Yao, G.; Gao, B.-M.; Fan, C.-X.; Bian, C.; Wang, J.; Cao, Y.; Wen, B.; Zhu, Y.; Ruan, Z.; et al. High-throughput identification of novel conotoxins from the Chinese tubular cone snail (Conus betulinus) by multitranscriptome sequencing. Gigascience 2016, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, S.D.; Li, Q.; Lu, A.; Bandyopadhyay, P.K.; Yandell, M.; Olivera, B.M.; Safavi-Hemami, H. The venom repertoire of Conus gloriamaris (Chemnitz, 1777), the glory of the sea. Mar. Drugs 2017, 15, 145. [Google Scholar] [CrossRef] [PubMed]
- Pardos-Blas, J.R.; Irisarri, I.; Abalde, S.; Tenorio, M.J.; Zardoya, R. Conotoxin diversity in the venom gland transcriptome of the magician’s cone, Pionoconus magus. Mar. Drugs 2019, 17, 553. [Google Scholar]
- Safavi-Hemami, H.; Siero, W.A.; Gorasia, D.G.; Young, N.D.; MacMillan, D.; Williamson, N.A.; Purcell, A.W. Specialisation of the venom gland proteome in predatory cone snails reveals functional diversification of the conotoxin biosynthetic pathway. J. Proteome Res. 2011, 10, 3904–3919. [Google Scholar] [CrossRef] [PubMed]
- Safavi-Hemami, H.; Lu, A.; Li, Q.; Fedosov, A.E.; Biggs, J.; Showers Corneli, P.; Seger, J.; Yandell, M.; Olivera, B.M. Venom Insulins of Cone Snails Diversify Rapidly and Track Prey Taxa. Mol. Biol. Evol. 2016, 33, 2924–2934. [Google Scholar] [CrossRef]
- Robinson, S.D.; Li, Q.; Bandyopadhyay, P.K.; Gajewiak, J.; Yandell, M.; Papenfuss, A.T.; Purcell, A.W.; Norton, R.S.; Safavi-Hemami, H. Hormone-like peptides in the venoms of marine cone snails. Gen. Comp. Endocrinol. 2017, 244, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, A.; Sajevic, T.; Pungerčar, J.; Križaj, I. Comprehensive study of the proteome and transcriptome of the venom of the most venomous european viper: Discovery of a new subclass of ancestral snake venom metalloproteinase precursor-derived proteins. J. Proteome Res. 2019, 18, 2287–2309. [Google Scholar] [CrossRef]
- Safavi-Hemami, H.; Hu, H.; Gorasia, D.G.; Bandyopadhyay, P.K.; Veith, P.D.; Young, N.D.; Reynolds, E.C.; Yandell, M.; Olivera, B.M.; Purcell, A.W. Combined proteomic and transcriptomic interrogation of the venom gland of Conus geographus uncovers novel components and functional compartmentalization. Mol. Cell. Proteom. 2014, 13, 938–953. [Google Scholar] [CrossRef] [Green Version]
- Violette, A.; Biass, D.; Dutertre, S.; Koua, D.; Piquemal, D.; Pierrat, F.; Stöcklin, R.; Favreau, P. Large-scale discovery of conopeptides and conoproteins in the injectable venom of a fish-hunting cone snail using a combined proteomic and transcriptomic approach. J. Proteom. 2012, 75, 5215–5225. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.; Vetter, I.; Himaya, S.W.A.; Alewood, P.F.; Lewis, R.J.; Dutertre, S. Transcriptome and proteome of Conus planorbis identify the nicotinic receptors as primary target for the defensive venom. Proteomics 2015, 15, 4030–4040. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Yu, R.; Jin, A.H.; Dutertre, S.; Craik, D.J. ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Barghi, N.; Lu, A.; Fedosov, E.A.; Bandyopadhyay, P.K.; Lluisma, O.A.; Concepcion, G.P.; Yandell, M.; Olivera, B.M.; Safavi-Hemami, H. Divergence of the Venom Exogene Repertoire in Two Sister Species of Turriconus. Genome Biol. Evol. 2017, 9, 2211–2225. [Google Scholar] [CrossRef] [Green Version]
- Möller, C.; Dovell, S.; Melaun, C.; Marí, F. Definition of the R-superfamily of conotoxins: Structural convergence of helix-loop-helix peptidic scaffolds. Peptides 2018, 107, 75–82. [Google Scholar] [CrossRef]
- Imperial, J.S.; Bansal, P.S.; Alewood, P.F.; Daly, N.L.; Craik, D.J.; Sporning, A.; Terlau, H.; López-Vera, E.; Bandyopadhyay, P.K.; Olivera, B.M. A novel conotoxin inhibitor of Kv1. 6 channel and nAChR subtypes defines a new superfamily of conotoxins. Biochemistry 2006, 45, 8331–8340. [Google Scholar] [CrossRef]
- Liu, Z.; Li, H.; Liu, N.; Wu, C.; Jiang, J.; Yue, J.; Jing, Y.; Dai, Q. Diversity and evolution of conotoxins in Conus virgo, Conus eburneus, Conus imperialis and Conus marmoreus from the South China Sea. Toxicon 2012, 60, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.D.; Safavi-Hemami, H.; McIntosh, L.D.; Purcell, A.W.; Norton, R.S.; Papenfuss, A.T. Diversity of Conotoxin Gene Superfamilies in the Venomous Snail, Conus victoriae. PLoS ONE 2014, 9, e87648. [Google Scholar]
- Smith, J.J.; Undheim, E.A.B. True lies: Using proteomics to assess the accuracy of transcriptome-based venomics in centipedes uncovers false positives and reveals startling intraspecific variation in Scolopendra subspinipes. Toxins 2018, 10, 96. [Google Scholar] [CrossRef] [Green Version]
- Endean, R.; Duchemin, C. The venom apparatus of Conus magus. Toxicon 1967, 4, 275–284. [Google Scholar] [CrossRef]
- Matsui, T.; Hamako, J. Structure and function of snake venom toxins interacting with human von Willebrand factor. Toxicon 2005, 45, 1075–1087. [Google Scholar] [CrossRef]
- Möller, C.; Davis, W.C.; Clark, E.; DeCaprio, A.; Marí, F. Conodipine-P1-3, the First Phospholipases A2 Characterized from Injected Cone Snail Venom. Mol. Cell. Proteom. 2019, 18, 876–891. [Google Scholar] [CrossRef]
- de Teixeira, C.F.P.; Landucci, E.C.T.; Antunes, E.; Chacur, M.; Cury, Y. Inflammatory effects of snake venom myotoxic phospholipases A2. Toxicon 2003, 42, 947–962. [Google Scholar] [CrossRef]
- Peiren, N.; de Graaf, D.C.; Vanrobaeys, F.; Danneels, E.L.; Devreese, B.; Van Beeumen, J.; Jacobs, F.J. Proteomic analysis of the honey bee worker venom gland focusing on the mechanisms of protection against tissue damage. Toxicon 2008, 52, 72–83. [Google Scholar] [CrossRef]
- Cruz, L.J.; Ramilo, C.A.; Corpuz, G.P.; Olivera, B.M. Conus peptides: Phylogenetic range of biological activity. Biol. Bull. 1992, 183, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Vijayasarathy, M.; Basheer, S.M.; Balaram, P. Cone snail glutaminyl cyclase sequences from transcriptomic analysis and mass spectrometric characterization of two pyroglutamyl conotoxins. J. Proteome Res. 2018, 17, 2695–2703. [Google Scholar] [CrossRef]
- Prator, C.A.; Murayama, K.M.; Schulz, R.J. Venom variation during prey capture by the cone snail, Conus textile. PLoS ONE 2014, 9, e98991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutertre, S.; Jin, A.H.; Kaas, Q.; Jones, A.; Alewood, P.F.; Lewis, R.J. Deep venomics reveals the mechanism for expanded peptide diversity in cone snail venom. Mol. Cell Proteom. 2013, 12, 312–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, J.K.; Tenorio, M.J. Illustrated Catalog of the Living Cone Shells; MdM Publishing: Wincanton, UK, 2013. [Google Scholar]
- Puillandre, N.; Duda, T.F.; Meyer, C.; Olivera, B.M.; Bouchet, P. One, four or 100 genera? A new classification of the cone snails. J. Molluscan Stud. 2015, 81, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abalde, S.; Tenorio, M.J.; Uribe, J.E.; Zardoya, R. Conidae phylogenomics and evolution. Zool. Scr. 2019, 48, 194–214. [Google Scholar] [CrossRef]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Calcott, B.; Ho, S.Y.W.; Guindon, S. Partitionfinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 7 August 2021).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Papanicolau, A. TransDecoder. 2010. Available online: https://github.com/TransDecoder/TransDecoder (accessed on 7 August 2021).
- Haas, B.J. Trinotate: Transcriptome Functional Annotation and Analysis. 2015. Available online: https://github.com/Trinotate/Trinotate.github.io/wiki (accessed on 7 August 2021).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.A.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using Tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Giribaldi, J.; Kazandjian, T.; Amorim, F.G.; Whiteley, G.; Wagstaff, S.C.; Cazals, G.; Enjalbal, C.; Quinton, L.; Casewell, N.R.; Dutertre, S. Venomics of the asp viper Vipera aspis aspis from France. J. Proteom. 2020, 218, 103707. [Google Scholar] [CrossRef]
- Lavergne, V.; Harliwong, I.; Jones, A.; Miller, D.; Taft, R.; Alewood, P.F. Optimized deep-targeted proteotranscriptomic profiling reveals unexplored Conus toxin diversity and novel cysteine frameworks. Proc. Natl. Acad. Sci. USA 2015, 112, E3782–E3791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavergne, V.; Dutertre, S.; Jin, A.-H.; Lewis, R.J.; Taft, R.J.; Alewood, P.F. Systematic interrogation of the Conus marmoreus venom duct transcriptome with ConoSorter reveals 158 novel conotoxins and 13 new gene superfamilies. BMC Genom. 2013, 14, 708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, B.; Peng, C.; Zhu, Y.; Sun, Y.; Zhao, T.; Huang, Y.; Shi, Q. High Throughput Identification of Novel Conotoxins from the Vermivorous Oak Cone Snail (Conus quercinus) by Transcriptome Sequencing. Int. J. Mol. Sci. 2018, 19, 3901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Team, R.C. R: A Language and Environment for Statistical Computing. 2013. Available online: https://www.r-project.org/ (accessed on 7 August 2021).
- Pusev, R.; Gavrilov, I. normtest: Tests for Normality. 2014. Available online: https://cran.r-project.org/web/packages/normtest/normtest.pdf (accessed on 7 August 2021).
- Dinno, A. Nonparametric pairwise multiple comparisons in independent groups using Dunn’s test. Stata J. 2015, 15, 292–300. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, R.A.M.; Chen, Z.J. ggplot2: Elegant Graphics for Data Analysis. 2019. Available online: https://ggplot2.tidyverse.org/ (accessed on 7 August 2021).




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abalde, S.; Dutertre, S.; Zardoya, R. A Combined Transcriptomics and Proteomics Approach Reveals the Differences in the Predatory and Defensive Venoms of the Molluscivorous Cone Snail Cylinder ammiralis (Caenogastropoda: Conidae). Toxins 2021, 13, 642. https://doi.org/10.3390/toxins13090642
Abalde S, Dutertre S, Zardoya R. A Combined Transcriptomics and Proteomics Approach Reveals the Differences in the Predatory and Defensive Venoms of the Molluscivorous Cone Snail Cylinder ammiralis (Caenogastropoda: Conidae). Toxins. 2021; 13(9):642. https://doi.org/10.3390/toxins13090642
Chicago/Turabian StyleAbalde, Samuel, Sébastien Dutertre, and Rafael Zardoya. 2021. "A Combined Transcriptomics and Proteomics Approach Reveals the Differences in the Predatory and Defensive Venoms of the Molluscivorous Cone Snail Cylinder ammiralis (Caenogastropoda: Conidae)" Toxins 13, no. 9: 642. https://doi.org/10.3390/toxins13090642
APA StyleAbalde, S., Dutertre, S., & Zardoya, R. (2021). A Combined Transcriptomics and Proteomics Approach Reveals the Differences in the Predatory and Defensive Venoms of the Molluscivorous Cone Snail Cylinder ammiralis (Caenogastropoda: Conidae). Toxins, 13(9), 642. https://doi.org/10.3390/toxins13090642