Clostridium perfringens Epsilon Toxin Binds to and Kills Primary Human Lymphocytes

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
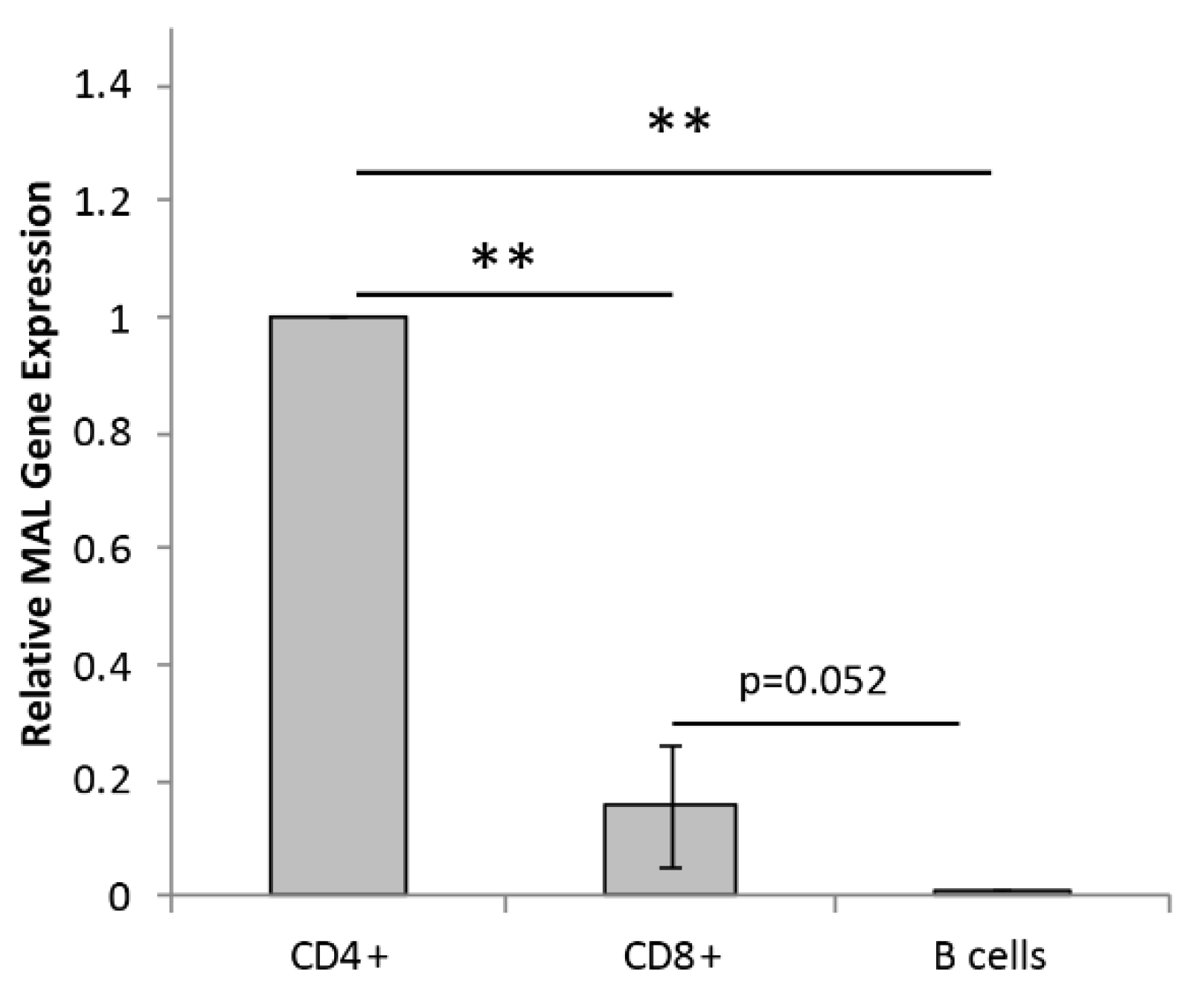
2.1. Primary Human Lymphocytes Express Mal
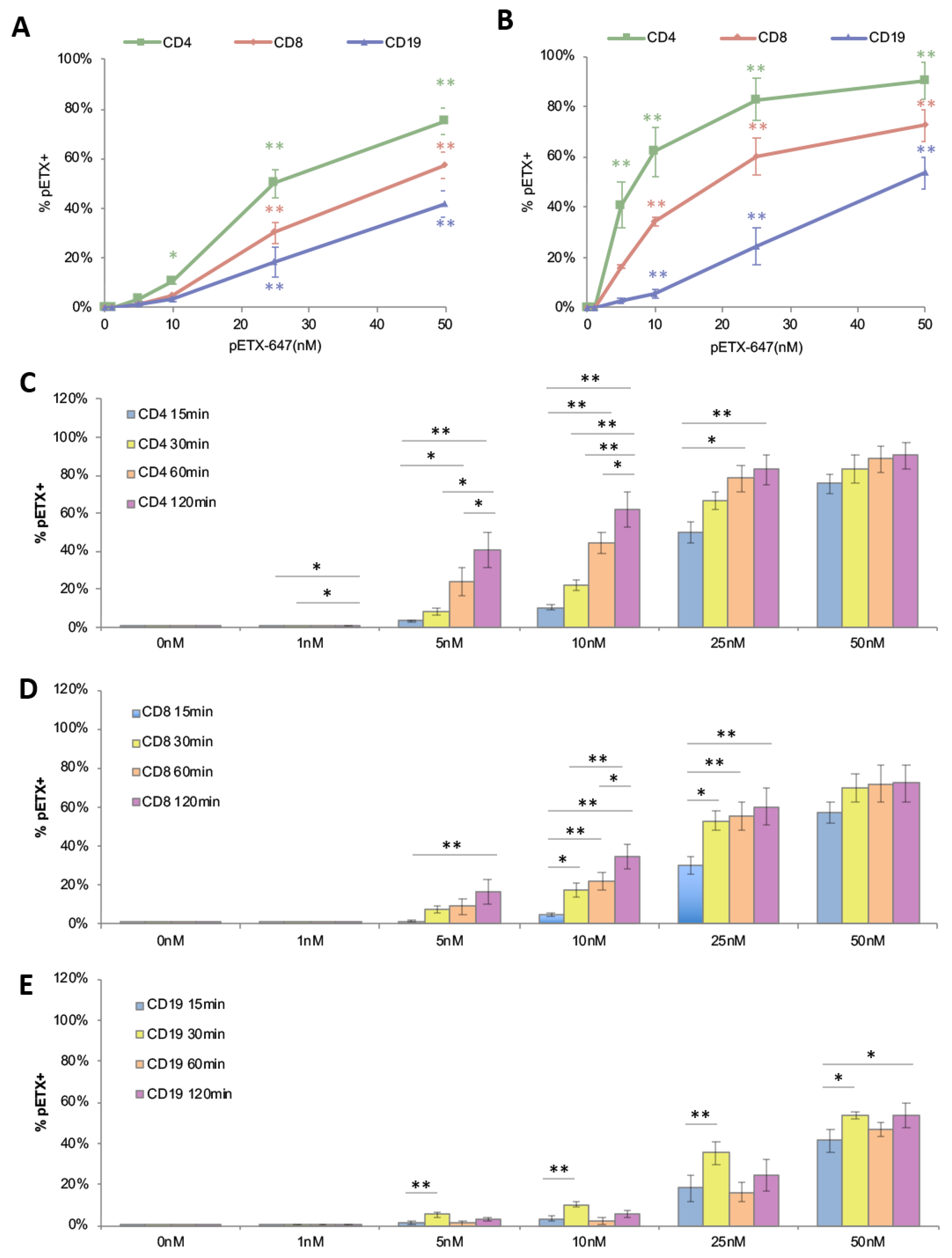
2.2. ETX Binds to Human Lymphocytes with a Preference for CD4+ Cells
2.3. ETX Bindings to Human Lymphocytes Is Dose and Time Dependent
2.4. ETX Induces Cytotoxicity in Human Lymphocytes, Especially CD4+ Cells
2.5. ETX-Induced Cytotoxicity in Human CD4+ Cells Is Time and Dose Dependent
2.6. ETX-Induced Cytotoxicity in Human Lymphocytes Is Mediated by Pore Formation
3. Discussion
3.1. ETX Binding and Cytotoxicity Positively Associates with Mal Gene Expression in Human Lymphocytes
3.2. MAL Expression in Human Lymphocytes
3.3. MAL’s Function in Different Lymphocyte Populations
3.4. ETX-Induced Cell Death Pathways
3.5. Possible Role of ETX-Lymphocytes Interactions in MS Pathogenesis
3.6. Conclusions
4. Materials and Methods
4.1. Peripheral Blood Isolation from Healthy Controls
4.2. Isolation of CD4+, CD8+, and B Cells from Human Peripheral Blood for RT-qPCR Analysis
4.3. Real-Time Quantitative PCR (RT-qPCR) Analysis
4.4. Preparation of Fluorescently Labeled pETX
4.5. Activation of ETX
4.6. PBMNC Isolation from Human Peripheral Blood for ETX Binding and Cytotoxicity Studies
4.7. Evaluation of pETX-647 Binding to Lymphocyte Subsets
4.8. Evaluation of ETX-Induced Cytotoxicity in Lymphocyte Subsets
4.9. Evalution of Pore Formation by Western Blot Analysis
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rumah, K.R.; Linden, J.; Fischetti, V.A.; Vartanian, T. Isolation of Clostridium perfringens type B in an individual at first clinical presentation of multiple sclerosis provides clues for environmental triggers of the disease. PLoS ONE 2013, 8, e76359. [Google Scholar] [CrossRef]
- Wagley, S.; Bokori-Brown, M.; Morcrette, H.; Malaspina, A.; D’Arcy, C.; Gnanapavan, S.; Lewis, N.; Popoff, M.R.; Raciborska, D.; Nicholas, R.; et al. Evidence of Clostridium perfringens epsilon toxin associated with multiple sclerosis. Mult. Scler. 2018, 25, 653–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Sannino, D.; Linden, J.R.; Haigh, S.; Zhao, B.; Grigg, J.B.; Zumbo, P.; Dundar, F.; Butler, D.; Profaci, C.P.; et al. Epsilon toxin-producing Clostridium perfringens colonize the multiple sclerosis gut microbiome overcoming CNS immune privilege. J. Clin. Investig. 2023, 133, e163239. [Google Scholar] [CrossRef] [PubMed]
- Adler, D.; Linden, J.R.; Shetty, S.V.; Ma, Y.; Bokori-Brown, M.; Titball, R.W.; Vartanian, T. Clostridium perfringens Epsilon Toxin Compromises the Blood-Brain Barrier in a Humanized Zebrafish Model. iScience 2019, 15, 39–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linden, J.R.; Flores, C.; Schmidt, E.F.; Uzal, F.A.; Michel, A.O.; Valenzuela, M.; Dobrow, S.; Vartanian, T. Clostridium perfringens epsilon toxin induces blood brain barrier permeability via caveolae-dependent transcytosis and requires expression of MAL. PLoS Pathog. 2019, 15, e1008014. [Google Scholar] [CrossRef] [Green Version]
- Linden, J.R.; Ma, Y.; Zhao, B.; Harris, J.M.; Rumah, K.R.; Schaeren-Wiemers, N.; Vartanian, T. Clostridium perfringens Epsilon Toxin Causes Selective Death of Mature Oligodendrocytes and Central Nervous System Demyelination. mBio 2015, 6, e02513. [Google Scholar] [CrossRef] [Green Version]
- Rumah, K.R.; Ma, Y.; Linden, J.R.; Oo, M.L.; Anrather, J.; Schaeren-Wiemers, N.; Alonso, M.A.; Fischetti, V.A.; McClain, M.S.; Vartanian, T. The Myelin and Lymphocyte Protein MAL Is Required for Binding and Activity of Clostridium perfringens epsilon-Toxin. PLoS Pathog. 2015, 11, e1004896. [Google Scholar] [CrossRef] [Green Version]
- Alonso, M.A.; Weissman, S.M. cDNA cloning and sequence of MAL, a hydrophobic protein associated with human T-cell differentiation. Proc. Natl. Acad. Sci. USA 1987, 84, 1997–2001. [Google Scholar] [CrossRef] [Green Version]
- Blanch, M.; Dorca-Arevalo, J.; Not, A.; Cases, M.; Gomez de Aranda, I.; Martinez Yelamos, A.; Martinez Yelamos, S.; Solsona, C.; Blasi, J. The cytotoxicity of Epsilon toxin from Clostridium perfringens on lymphocytes is mediated by MAL protein expression. Mol. Cell. Biol. 2018, 38, e00086-18. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Zhang, P.; Wang, J.; Ma, L.; E, W.; Suo, S.; Jiang, M.; Li, J.; Chen, H.; Sun, H.; et al. Construction of a cross-species cell landscape at single-cell level. Nucleic Acids Res. 2023, 51, 501–516. [Google Scholar] [CrossRef]
- van Langelaar, J.; Rijvers, L.; Smolders, J.; van Luijn, M.M. B and T Cells Driving Multiple Sclerosis: Identity, Mechanisms and Potential Triggers. Front. Immunol. 2020, 11, 760. [Google Scholar] [CrossRef]
- Chitnis, T. The role of CD4 T cells in the pathogenesis of multiple sclerosis. Int. Rev. Neurobiol. 2007, 79, 43–72. [Google Scholar] [PubMed]
- Bokori-Brown, M.; Savva, C.G.; Fernandes da Costa, S.P.; Naylor, C.E.; Basak, A.K.; Titball, R.W. Molecular basis of toxicity of Clostridium perfringens epsilon toxin. FEBS J. 2011, 278, 4589–4601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popoff, M.R. Epsilon toxin: A fascinating pore-forming toxin. FEBS J. 2011, 278, 4602–4615. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.L.; Li, J.; Uzal, F.A.; McClane, B.A. Evidence for a prepore stage in the action of Clostridium perfringens epsilon toxin. PLoS ONE 2011, 6, e22053. [Google Scholar] [CrossRef] [Green Version]
- Savva, C.G.; Clark, A.R.; Naylor, C.E.; Popoff, M.R.; Moss, D.S.; Basak, A.K.; Titball, R.W.; Bokori-Brown, M. The pore structure of Clostridium perfringens epsilon toxin. Nat. Commun. 2019, 10, 2641. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.P.; Adams, V.; Beingesser, J.; Hughes, M.L.; Poon, R.; Lyras, D.; Hill, A.; McClane, B.A.; Rood, J.I.; Uzal, F.A. Epsilon toxin is essential for the virulence of Clostridium perfringens type D infection in sheep, goats, and mice. Infect. Immun. 2013, 81, 2405–2414. [Google Scholar] [CrossRef] [Green Version]
- Uzal, F.A.; Pasini, M.I.; Olaechea, F.V.; Robles, C.A.; Elizondo, A. An outbreak of enterotoxaemia caused by Clostridium perfringens type D in goats in Patagonia. Vet. Rec. 1994, 135, 279–280. [Google Scholar] [CrossRef]
- Nagahama, M.; Sakurai, J. Distribution of labeled Clostridium perfringens epsilon toxin in mice. Toxicon 1991, 29, 211–217. [Google Scholar] [CrossRef]
- Tamai, E.; Ishida, T.; Miyata, S.; Matsushita, O.; Suda, H.; Kobayashi, S.; Sonobe, H.; Okabe, A. Accumulation of Clostridium perfringens epsilon-toxin in the mouse kidney and its possible biological significance. Infect. Immun. 2003, 71, 5371–5375. [Google Scholar] [CrossRef] [Green Version]
- Finnie, J.W. Pathogenesis of brain damage produced in sheep by Clostridium perfringens type D epsilon toxin: A review. Aust. Vet. J. 2003, 81, 219–221. [Google Scholar] [CrossRef]
- Soler-Jover, A.; Blasi, J.; Gomez de Aranda, I.; Navarro, P.; Gibert, M.; Popoff, M.R.; Martin-Satue, M. Effect of epsilon toxin-GFP on MDCK cells and renal tubules in vivo. J. Histochem. Cytochem. 2004, 52, 931–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez Miyakawa, M.E.; Zabal, O.; Silberstein, C. Clostridium perfringens epsilon toxin is cytotoxic for human renal tubular epithelial cells. Hum. Exp. Toxicol. 2011, 30, 275–282. [Google Scholar] [CrossRef]
- Dorca-Arevalo, J.; Martin-Satue, M.; Blasi, J. Characterization of the high affinity binding of epsilon toxin from Clostridium perfringens to the renal system. Vet. Microbiol. 2012, 157, 179–189. [Google Scholar] [CrossRef]
- Finnie, J.W. Neurological disorders produced by Clostridium perfringens type D epsilon toxin. Anaerobe 2004, 10, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Dorca-Arevalo, J.; Soler-Jover, A.; Gibert, M.; Popoff, M.R.; Martin-Satue, M.; Blasi, J. Binding of epsilon-toxin from Clostridium perfringens in the nervous system. Vet. Microbiol. 2008, 131, 14–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wioland, L.; Dupont, J.L.; Doussau, F.; Gaillard, S.; Heid, F.; Isope, P.; Pauillac, S.; Popoff, M.R.; Bossu, J.L.; Poulain, B. Epsilon toxin from Clostridium perfringens acts on oligodendrocytes without forming pores, and causes demyelination. Cell. Microbiol. 2015, 17, 369–388. [Google Scholar] [CrossRef] [Green Version]
- Lonchamp, E.; Dupont, J.L.; Wioland, L.; Courjaret, R.; Mbebi-Liegeois, C.; Jover, E.; Doussau, F.; Popoff, M.R.; Bossu, J.L.; de Barry, J.; et al. Clostridium perfringens epsilon toxin targets granule cells in the mouse cerebellum and stimulates glutamate release. PLoS ONE 2010, 5, e13046. [Google Scholar] [CrossRef]
- Worthington, R.W.; Mulders, M.S. Effect of Clostridium perfringens epsilon toxin on the blood brain barrier of mice. Onderstepoort J. Vet. Res. 1975, 42, 25–27. [Google Scholar]
- Soler-Jover, A.; Dorca, J.; Popoff, M.R.; Gibert, M.; Saura, J.; Tusell, J.M.; Serratosa, J.; Blasi, J.; Martin-Satue, M. Distribution of Clostridium perfringens epsilon toxin in the brains of acutely intoxicated mice and its effect upon glial cells. Toxicon 2007, 50, 530–540. [Google Scholar] [CrossRef]
- Fennessey, C.M.; Sheng, J.; Rubin, D.H.; McClain, M.S. Oligomerization of Clostridium perfringens epsilon toxin is dependent upon caveolins 1 and 2. PLoS ONE 2012, 7, e46866. [Google Scholar] [CrossRef] [PubMed]
- Ivie, S.E.; Fennessey, C.M.; Sheng, J.; Rubin, D.H.; McClain, M.S. Gene-trap mutagenesis identifies mammalian genes contributing to intoxication by Clostridium perfringens epsilon-toxin. PLoS ONE 2011, 6, e17787. [Google Scholar] [CrossRef] [PubMed]
- Anton, O.; Batista, A.; Millan, J.; Andres-Delgado, L.; Puertollano, R.; Correas, I.; Alonso, M.A. An essential role for the MAL protein in targeting Lck to the plasma membrane of human T lymphocytes. J. Exp. Med. 2008, 205, 3201–3213. [Google Scholar] [CrossRef] [Green Version]
- Anton, O.M.; Andres-Delgado, L.; Reglero-Real, N.; Batista, A.; Alonso, M.A. MAL protein controls protein sorting at the supramolecular activation cluster of human T lymphocytes. J. Immunol. 2011, 186, 6345–6356. [Google Scholar] [CrossRef] [Green Version]
- Millan, J.; Alonso, M.A. MAL, a novel integral membrane protein of human T lymphocytes, associates with glycosylphosphatidylinositol-anchored proteins and Src-like tyrosine kinases. Eur. J. Immunol. 1998, 28, 3675–3684. [Google Scholar] [CrossRef]
- Millan, J.; Puertollano, R.; Fan, L.; Alonso, M.A. Caveolin and MAL, two protein components of internal detergent-insoluble membranes, are in distinct lipid microenvironments in MDCK cells. Biochem. Biophys. Res. Commun. 1997, 233, 707–712. [Google Scholar] [CrossRef]
- Millan, J.; Puertollano, R.; Fan, L.; Rancano, C.; Alonso, M.A. The MAL proteolipid is a component of the detergent-insoluble membrane subdomains of human T-lymphocytes. Biochem. J. 1997, 321 Pt 1, 247–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventimiglia, L.N.; Fernandez-Martin, L.; Martinez-Alonso, E.; Anton, O.M.; Guerra, M.; Martinez-Menarguez, J.A.; Andres, G.; Alonso, M.A. Cutting Edge: Regulation of Exosome Secretion by the Integral MAL Protein in T Cells. J. Immunol. 2015, 195, 810–814. [Google Scholar] [CrossRef] [Green Version]
- Frank, M. MAL, a proteolipid in glycosphingolipid enriched domains: Functional implications in myelin and beyond. Prog. Neurobiol. 2000, 60, 531–544. [Google Scholar] [CrossRef]
- Schaeren-Wiemers, N.; Schaefer, C.; Valenzuela, D.M.; Yancopoulos, G.D.; Schwab, M.E. Identification of new oligodendrocyte- and myelin-specific genes by a differential screening approach. J. Neurochem. 1995, 65, 10–22. [Google Scholar] [CrossRef]
- Schaeren-Wiemers, N.; Valenzuela, D.M.; Frank, M.; Schwab, M.E. Characterization of a rat gene, rMAL, encoding a protein with four hydrophobic domains in central and peripheral myelin. J. Neurosci. 1995, 15, 5753–5764. [Google Scholar] [CrossRef] [PubMed]
- Zacchetti, D.; Peranen, J.; Murata, M.; Fiedler, K.; Simons, K. VIP17/MAL, a proteolipid in apical transport vesicles. FEBS Lett. 1995, 377, 465–469. [Google Scholar] [PubMed] [Green Version]
- Kim, T.; Fiedler, K.; Madison, D.L.; Krueger, W.H.; Pfeiffer, S.E. Cloning and characterization of MVP17: A developmentally regulated myelin protein in oligodendrocytes. J. Neurosci. Res. 1995, 42, 413–422. [Google Scholar] [CrossRef]
- Caduff, J.; Sansano, S.; Bonnet, A.; Suter, U.; Schaeren-Wiemers, N. Characterization of GFP-MAL expression and incorporation in rafts. Microsc. Res. Tech. 2001, 52, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Magal, L.G.; Yaffe, Y.; Shepshelovich, J.; Aranda, J.F.; de Marco Mdel, C.; Gaus, K.; Alonso, M.A.; Hirschberg, K. Clustering and lateral concentration of raft lipids by the MAL protein. Mol. Biol. Cell 2009, 20, 3751–3762. [Google Scholar] [CrossRef] [Green Version]
- Ramnarayanan, S.P.; Tuma, P.L. MAL, but not MAL2, expression promotes the formation of cholesterol-dependent membrane domains that recruit apical proteins. Biochem. J. 2011, 439, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Alonso, M.A.; Millan, J. The role of lipid rafts in signalling and membrane trafficking in T lymphocytes. J. Cell. Sci. 2001, 114 Pt 22, 3957–3965. [Google Scholar] [CrossRef]
- Martin-Belmonte, F.; Arvan, P.; Alonso, M.A. MAL mediates apical transport of secretory proteins in polarized epithelial Madin-Darby canine kidney cells. J. Biol. Chem. 2001, 276, 49337–49342. [Google Scholar] [CrossRef] [Green Version]
- Martin-Belmonte, F.; Puertollano, R.; Millan, J.; Alonso, M.A. The MAL proteolipid is necessary for the overall apical delivery of membrane proteins in the polarized epithelial Madin-Darby canine kidney and fischer rat thyroid cell lines. Mol. Biol. Cell 2000, 11, 2033–2045. [Google Scholar] [CrossRef] [Green Version]
- Puertollano, R.; Alonso, M.A. MAL, an integral element of the apical sorting machinery, is an itinerant protein that cycles between the trans-Golgi network and the plasma membrane. Mol. Biol. Cell 1999, 10, 3435–3447. [Google Scholar] [CrossRef] [Green Version]
- Puertollano, R.; Martin-Belmonte, F.; Millan, J.; de Marco, M.C.; Albar, J.P.; Kremer, L.; Alonso, M.A. The MAL proteolipid is necessary for normal apical transport and accurate sorting of the influenza virus hemagglutinin in Madin-Darby canine kidney cells. J. Cell Biol. 1999, 145, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puertollano, R.; Martinez-Menarguez, J.A.; Batista, A.; Ballesta, J.; Alonso, M.A. An intact dilysine-like motif in the carboxyl terminus of MAL is required for normal apical transport of the influenza virus hemagglutinin cargo protein in epithelial Madin-Darby canine kidney cells. Mol. Biol. Cell 2001, 12, 1869–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puertollano, R.; Menendez, M.; Alonso, M.A. Incorporation of MAL, an integral protein element of the machinery for the glycolipid and cholesterol-mediated apical pathway of transport, into artificial membranes requires neither of these lipid species. Biochem. Biophys. Res. Commun. 1999, 266, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Schaeren-Wiemers, N.; Bonnet, A.; Erb, M.; Erne, B.; Bartsch, U.; Kern, F.; Mantei, N.; Sherman, D.; Suter, U. The raft-associated protein MAL is required for maintenance of proper axon--glia interactions in the central nervous system. J. Cell Biol. 2004, 166, 731–742. [Google Scholar] [CrossRef] [Green Version]
- Andres-Delgado, L.; Anton, O.M.; Madrid, R.; Byrne, J.A.; Alonso, M.A. Formin INF2 regulates MAL-mediated transport of Lck to the plasma membrane of human T lymphocytes. Blood 2010, 116, 5919–5929. [Google Scholar] [CrossRef] [Green Version]
- Copie-Bergman, C.; Plonquet, A.; Alonso, M.A.; Boulland, M.L.; Marquet, J.; Divine, M.; Moller, P.; Leroy, K.; Gaulard, P. MAL expression in lymphoid cells: Further evidence for MAL as a distinct molecular marker of primary mediastinal large B-cell lymphomas. Mod. Pathol. 2002, 15, 1172–1180. [Google Scholar] [CrossRef] [Green Version]
- Rancano, C.; Rubio, T.; Alonso, M.A. Alternative splicing of human T-cell-specific MAL mRNA and its correlation with the exon/intron organization of the gene. Genomics 1994, 21, 447–450. [Google Scholar] [CrossRef]
- Rancano, C.; Rubio, T.; Correas, I.; Alonso, M.A. Genomic structure and subcellular localization of MAL, a human T-cell-specific proteolipid protein. J. Biol. Chem. 1994, 269, 8159–8164. [Google Scholar] [CrossRef]
- Gentry, M.; Bodo, J.; Durkin, L.; Hsi, E.D. Performance of a Commercially Available MAL Antibody in the Diagnosis of Primary Mediastinal Large B-Cell Lymphoma. Am. J. Surg. Pathol. 2017, 41, 189–194. [Google Scholar] [CrossRef]
- Kohno, T.; Moriuchi, R.; Katamine, S.; Yamada, Y.; Tomonaga, M.; Matsuyama, T. Identification of genes associated with the progression of adult T cell leukemia (ATL). Jpn. J. Cancer Res. 2000, 91, 1103–1110. [Google Scholar] [CrossRef]
- Lara-Lemus, R. On The Role of Myelin and Lymphocyte Protein (MAL) In Cancer: A Puzzle With Two Faces. J. Cancer 2019, 10, 2312–2318. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Ramos, A.; Labat-de-Hoz, L.; Correas, I.; Alonso, M.A. The MAL Protein, an Integral Component of Specialized Membranes, in Normal Cells and Cancer. Cells 2021, 10, 1065. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K. Mediastinal large B-cell lymphoma: New evidence in support of its distinctive identity. Adv. Anat. Pathol. 2000, 7, 201–209. [Google Scholar] [CrossRef]
- Copie-Bergman, C.; Gaulard, P.; Maouche-Chretien, L.; Briere, J.; Haioun, C.; Alonso, M.A.; Romeo, P.H.; Leroy, K. The MAL gene is expressed in primary mediastinal large B-cell lymphoma. Blood 1999, 94, 3567–3575. [Google Scholar] [CrossRef] [PubMed]
- Hsi, E.D.; Sup, S.J.; Alemany, C.; Tso, E.; Skacel, M.; Elson, P.; Alonso, M.A.; Pohlman, B. MAL is expressed in a subset of Hodgkin lymphoma and identifies a population of patients with poor prognosis. Am. J. Clin. Pathol. 2006, 125, 776–782. [Google Scholar] [CrossRef]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlen, M.; Karlsson, M.J.; Zhong, W.; Tebani, A.; Pou, C.; Mikes, J.; Lakshmikanth, T.; Forsstrom, B.; Edfors, F.; Odeberg, J.; et al. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science 2019, 366, eaax9198. [Google Scholar] [CrossRef]
- Watkins, N.A.; Gusnanto, A.; de Bono, B.; De, S.; Miranda-Saavedra, D.; Hardie, D.L.; Angenent, W.G.; Attwood, A.P.; Ellis, P.D.; Erber, W.; et al. A HaemAtlas: Characterizing gene expression in differentiated human blood cells. Blood 2009, 113, e1–e9. [Google Scholar] [CrossRef]
- Choi, J.; Baldwin, T.M.; Wong, M.; Bolden, J.E.; Fairfax, K.A.; Lucas, E.C.; Cole, R.; Biben, C.; Morgan, C.; Ramsay, K.A.; et al. Haemopedia RNA-seq: A database of gene expression during haematopoiesis in mice and humans. Nucleic Acids Res. 2019, 47, D780–D785. [Google Scholar] [CrossRef] [Green Version]
- Nagahama, M.; Ochi, S.; Sakurai, J. Assembly of Clostridium perfringens epsilon-toxin on MDCK cell membrane. J. Nat. Toxins 1998, 7, 291–302. [Google Scholar]
- Petit, L.; Gibert, M.; Gillet, D.; Laurent-Winter, C.; Boquet, P.; Popoff, M.R. Clostridium perfringens epsilon-toxin acts on MDCK cells by forming a large membrane complex. J. Bacteriol. 1997, 179, 6480–6487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorca-Arevalo, J.; Gomez de Aranda, I.; Blasi, J. New Mutants of Epsilon Toxin from Clostridium perfringens with an Altered Receptor-Binding Site and Cell-Type Specificity. Toxins 2022, 14, 288. [Google Scholar] [CrossRef]
- Miyata, S.; Matsushita, O.; Minami, J.; Katayama, S.; Shimamoto, S.; Okabe, A. Cleavage of a C-terminal peptide is essential for heptamerization of Clostridium perfringens epsilon-toxin in the synaptosomal membrane. J. Biol. Chem. 2001, 276, 13778–13783. [Google Scholar] [CrossRef] [Green Version]
- Miyata, S.; Minami, J.; Tamai, E.; Matsushita, O.; Shimamoto, S.; Okabe, A. Clostridium perfringens epsilon-toxin forms a heptameric pore within the detergent-insoluble microdomains of Madin-Darby canine kidney cells and rat synaptosomes. J. Biol. Chem. 2002, 277, 39463–39468. [Google Scholar] [CrossRef] [Green Version]
- Llorente, A.; de Marco, M.C.; Alonso, M.A. Caveolin-1 and MAL are located on prostasomes secreted by the prostate cancer PC-3 cell line. J. Cell Sci. 2004, 117 Pt 22, 5343–5351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagahama, M.; Itohayashi, Y.; Hara, H.; Higashihara, M.; Fukatani, Y.; Takagishi, T.; Oda, M.; Kobayashi, K.; Nakagawa, I.; Sakurai, J. Cellular vacuolation induced by Clostridium perfringens epsilon-toxin. FEBS J. 2011, 278, 3395–3407. [Google Scholar] [CrossRef] [PubMed]
- Ventimiglia, L.N.; Alonso, M.A. Biogenesis and Function of T Cell-Derived Exosomes. Front. Cell Dev. Biol. 2016, 4, 84. [Google Scholar] [CrossRef] [Green Version]
- Linden, J.R.; Telesford, K.; Shetty, S.; Winokour, P.; Haigh, S.; Cahir-McFarland, E.; Antognetti, G.; Datta, A.; Wang, T.; Meier, W.; et al. A Novel Panel of Rabbit Monoclonal Antibodies and Their Diverse Applications Including Inhibition of Clostridium perfringens Epsilon Toxin Oligomerization. Antibodies 2018, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Kouzel, I.U.; Pohlentz, G.; Storck, W.; Radamm, L.; Hoffmann, P.; Bielaszewska, M.; Bauwens, A.; Cichon, C.; Schmidt, M.A.; Mormann, M.; et al. Association of Shiga toxin glycosphingolipid receptors with membrane microdomains of toxin-sensitive lymphoid and myeloid cells. J. Lipid Res. 2013, 54, 692–710. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.I.; Czarnecka, A.M.; Lewicki, S.; Helbrecht, I.; Brodaczewska, K.; Koch, I.; Zdanowski, R.; Krol, M.; Szczylik, C. Comparative Gene Expression Profiling of Primary and Metastatic Renal Cell Carcinoma Stem Cell-Like Cancer Cells. PLoS ONE 2016, 11, e0165718. [Google Scholar] [CrossRef] [Green Version]
- Martin-Belmonte, F.; Kremer, L.; Albar, J.P.; Marazuela, M.; Alonso, M.A. Expression of the MAL gene in the thyroid: The MAL proteolipid, a component of glycolipid-enriched membranes, is apically distributed in thyroid follicles. Endocrinology 1998, 139, 2077–2084. [Google Scholar] [CrossRef]
- Palacios, E.H.; Weiss, A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 2004, 23, 7990–8000. [Google Scholar] [CrossRef] [Green Version]
- Pierce, S.K. Lipid rafts and B-cell activation. Nat. Rev. Immunol. 2002, 2, 96–105. [Google Scholar] [CrossRef]
- Dorfman, D.M.; Shahsafaei, A.; Alonso, M.A. Utility of CD200 immunostaining in the diagnosis of primary mediastinal large B cell lymphoma: Comparison with MAL, CD23, and other markers. Mod. Pathol. 2012, 25, 1637–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petit, L.; Gibert, M.; Gourch, A.; Bens, M.; Vandewalle, A.; Popoff, M.R. Clostridium perfringens epsilon toxin rapidly decreases membrane barrier permeability of polarized MDCK cells. Cell. Microbiol. 2003, 5, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Chassin, C.; Bens, M.; de Barry, J.; Courjaret, R.; Bossu, J.L.; Cluzeaud, F.; Ben Mkaddem, S.; Gibert, M.; Poulain, B.; Popoff, M.R.; et al. Pore-forming epsilon toxin causes membrane permeabilization and rapid ATP depletion-mediated cell death in renal collecting duct cells. Am. J. Physiol. Renal Physiol. 2007, 293, F927–F937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.; Gao, J.; Yao, W.; Kang, L.; Gao, S.; Yang, H.; Ji, B.; Li, P.; Liu, J.; Yao, J.; et al. F199E substitution reduced toxicity of Clostridium perfringens epsilon toxin by depriving the receptor binding capability. Hum. Vaccines Immunother. 2017, 13, 1598–1608. [Google Scholar] [CrossRef] [Green Version]
- Frischer, J.M.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Bruck, W.; et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann. Neurol. 2015, 78, 710–721. [Google Scholar] [CrossRef] [Green Version]
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Bruck, W.; Lassmann, H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017, 133, 13–24. [Google Scholar] [CrossRef]
- Ramaglia, V.; Sheikh-Mohamed, S.; Legg, K.; Park, C.; Rojas, O.L.; Zandee, S.; Fu, F.; Ornatsky, O.; Swanson, E.C.; Pitt, D.; et al. Multiplexed imaging of immune cells in staged multiple sclerosis lesions by mass cytometry. Elife 2019, 8, e48051. [Google Scholar] [CrossRef]
- Sospedra, M.; Martin, R. Immunology of Multiple Sclerosis. Semin. Neurol. 2016, 36, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucchinetti, C.; Bruck, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Bischof, L.J.; Kao, C.Y.; Los, F.C.; Gonzalez, M.R.; Shen, Z.; Briggs, S.P.; van der Goot, F.G.; Aroian, R.V. Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog. 2008, 4, e1000176. [Google Scholar] [CrossRef] [PubMed]
- Bischofberger, M.; Iacovache, I.; van der Goot, F.G. Pathogenic pore-forming proteins: Function and host response. Cell Host Microbe 2012, 12, 266–275. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.S.; Bellier, A.; Kao, C.Y.; Yang, Y.L.; Chen, H.D.; Los, F.C.; Aroian, R.V. WWP-1 is a novel modulator of the DAF-2 insulin-like signaling network involved in pore-forming toxin cellular defenses in Caenorhabditis elegans. PLoS ONE 2010, 5, e9494. [Google Scholar] [CrossRef]
- Gonzalez, M.R.; Bischofberger, M.; Freche, B.; Ho, S.; Parton, R.G.; van der Goot, F.G. Pore-forming toxins induce multiple cellular responses promoting survival. Cell. Microbiol. 2011, 13, 1026–1043. [Google Scholar] [CrossRef] [Green Version]
- Gurcel, L.; Abrami, L.; Girardin, S.; Tschopp, J.; van der Goot, F.G. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 2006, 126, 1135–1145. [Google Scholar] [CrossRef] [Green Version]
- Huffman, D.L.; Abrami, L.; Sasik, R.; Corbeil, J.; van der Goot, F.G.; Aroian, R.V. Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc. Natl. Acad. Sci. USA 2004, 101, 10995–11000. [Google Scholar] [CrossRef] [Green Version]
- Kao, C.Y.; Los, F.C.; Huffman, D.L.; Wachi, S.; Kloft, N.; Husmann, M.; Karabrahimi, V.; Schwartz, J.L.; Bellier, A.; Ha, C.; et al. Global functional analyses of cellular responses to pore-forming toxins. PLoS Pathog. 2011, 7, e1001314. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, C.L.; Smith, D.J.; Lyras, D.; Chakravorty, A.; Rood, J.I. Programmed cellular necrosis mediated by the pore-forming alpha-toxin from Clostridium septicum. PLoS Pathog. 2009, 5, e1000516. [Google Scholar] [CrossRef] [Green Version]
- Wiles, T.J.; Dhakal, B.K.; Eto, D.S.; Mulvey, M.A. Inactivation of host Akt/protein kinase B signaling by bacterial pore-forming toxins. Mol. Biol. Cell 2008, 19, 1427–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, P.; Gandhi, S.; Lata, K.; Chattopadhyay, K. Pore-forming toxins in infection and immunity. Biochem. Soc. Trans. 2021, 49, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shetty, S.V.; Mazzucco, M.R.; Winokur, P.; Haigh, S.V.; Rumah, K.R.; Fischetti, V.A.; Vartanian, T.; Linden, J.R. Clostridium perfringens Epsilon Toxin Binds to and Kills Primary Human Lymphocytes. Toxins 2023, 15, 423. https://doi.org/10.3390/toxins15070423
Shetty SV, Mazzucco MR, Winokur P, Haigh SV, Rumah KR, Fischetti VA, Vartanian T, Linden JR. Clostridium perfringens Epsilon Toxin Binds to and Kills Primary Human Lymphocytes. Toxins. 2023; 15(7):423. https://doi.org/10.3390/toxins15070423
Chicago/Turabian StyleShetty, Samantha V., Michael R. Mazzucco, Paige Winokur, Sylvia V. Haigh, Kareem Rashid Rumah, Vincent A. Fischetti, Timothy Vartanian, and Jennifer R. Linden. 2023. "Clostridium perfringens Epsilon Toxin Binds to and Kills Primary Human Lymphocytes" Toxins 15, no. 7: 423. https://doi.org/10.3390/toxins15070423
APA StyleShetty, S. V., Mazzucco, M. R., Winokur, P., Haigh, S. V., Rumah, K. R., Fischetti, V. A., Vartanian, T., & Linden, J. R. (2023). Clostridium perfringens Epsilon Toxin Binds to and Kills Primary Human Lymphocytes. Toxins, 15(7), 423. https://doi.org/10.3390/toxins15070423