Recent Developments in Vaccine Design: From Live Vaccines to Recombinant Toxin Vaccines


Abstract
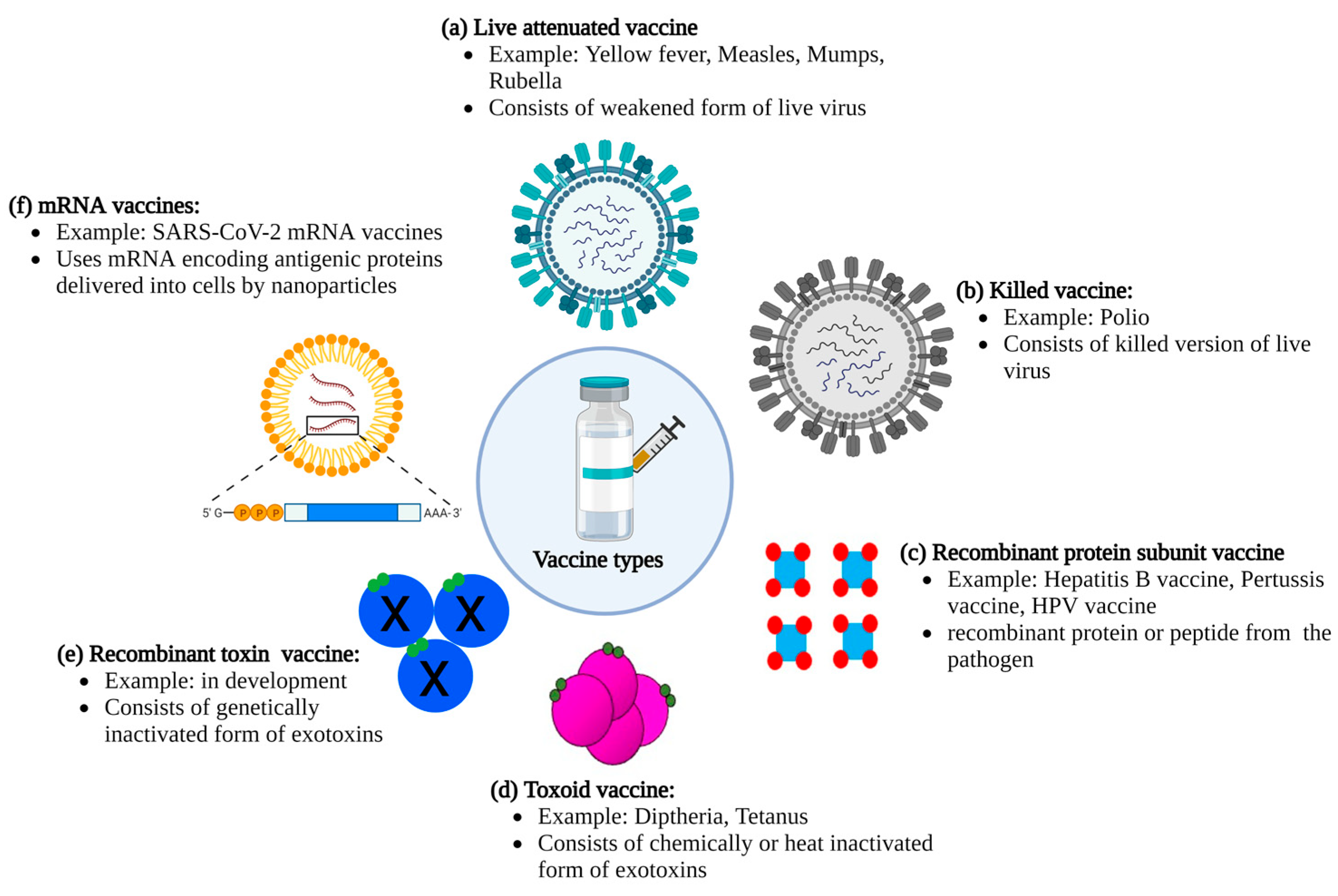
:1. Introduction
2. Live Attenuated Vaccines
3. Killed Vaccines
4. Toxoid Vaccine
4.1. Tetanus Toxoid Vaccines
4.2. Diphtheria Toxoid Vaccines
4.3. Botulinum Neurotoxin Toxoid Vaccines
5. Recombinant Subunit and Toxin Vaccines
5.1. Recombinant Tetanus Toxin Vaccines
5.2. Recombinant Botulinum Neurotoxin Vaccines
5.3. Recombinant Subunit Vaccines to SARS-CoV-2
6. mRNA Vaccines
6.1. SARS-CoV-2 mRNA Vaccines
6.2. Developments in mRNA Immunotherapy to Botulinum Neurotoxins
7. Discussion and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Melief, C.J.; van Hall, T.; Arens, R.; Ossendorp, F.; van der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef]
- Bol, K.F.; Aarntzen, E.H.; Pots, J.M.; Olde Nordkamp, M.A.; van de Rakt, M.W.; Scharenborg, N.M.; de Boer, A.J.; van Oorschot, T.G.; Croockewit, S.A.; Blokx, W.A.; et al. Prophylactic vaccines are potent activators of monocyte-derived dendritic cells and drive effective anti-tumor responses in melanoma patients at the cost of toxicity. Cancer Immunol. Immunother. 2016, 65, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Riedel, S. Edward Jenner and the history of smallpox and vaccination. In Baylor University Medical Center Proceedings; Taylor & Francis: Oxford, UK, 2005; Volume 18, pp. 21–25. [Google Scholar] [CrossRef]
- Plotkin, S. History of vaccination. Proc. Natl. Acad. Sci. USA 2014, 111, 12283–12287. [Google Scholar] [CrossRef] [PubMed]
- Nabel, G.J. Designing tomorrow’s vaccines. N. Engl. J. Med. 2013, 368, 551–560. [Google Scholar] [CrossRef]
- Plotkin, S.A. Vaccination in the 21st century. J. Infect. Dis. 1993, 168, 29–37. [Google Scholar] [CrossRef]
- Plotkin, S.A. Vaccines: Past, present and future. Nat. Med. 2005, 11, S5–S11. [Google Scholar] [CrossRef]
- Clark, T.G.; Cassidy-Hanley, D. Recombinant subunit vaccines: Potentials and constraints. Dev. Biol. 2005, 121, 153–163. [Google Scholar]
- Nascimento, I.P.; Leite, L.C. Recombinant vaccines and the development of new vaccine strategies. Braz. J. Med. Biol. Res. 2012, 45, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Simborio, H.L.; Lee, J.J.; Bernardo Reyes, A.W.; Hop, H.T.; Arayan, L.T.; Min, W.; Lee, H.J.; Yoo, H.S.; Kim, S. Evaluation of the combined use of the recombinant Brucella abortus Omp10, Omp19 and Omp28 proteins for the clinical diagnosis of bovine brucellosis. Microb. Pathog. 2015, 83–84, 41–46. [Google Scholar] [CrossRef]
- Hotez, P.J.; Bottazzi, M.E. Whole Inactivated Virus and Protein-Based COVID-19 Vaccines. Annu. Rev. Med. 2022, 73, 55–64. [Google Scholar] [CrossRef]
- Lehrer, A.T.; Chuang, E.; Namekar, M.; Williams, C.A.; Wong, T.A.S.; Lieberman, M.M.; Granados, A.; Misamore, J.; Yalley-Ogunro, J.; Andersen, H.; et al. Recombinant Protein Filovirus Vaccines Protect Cynomolgus Macaques from Ebola, Sudan, and Marburg Viruses. Front. Immunol. 2021, 12, 703986. [Google Scholar] [CrossRef]
- Prygiel, M.; Mosiej, E.; Gorska, P.; Zasada, A.A. Diphtheria-tetanus-pertussis vaccine: Past, current & future. Future Microbiol. 2022, 17, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.W. Diphtheria and tetanus toxoids. Br. Med. Bull. 1969, 25, 177–182. [Google Scholar] [CrossRef]
- Przedpelski, A.; Tepp, W.H.; Pellett, S.; Johnson, E.A.; Barbieri, J.T. A Novel High-Potency Tetanus Vaccine. mBio 2020, 11, 01668-20. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.; Thorp, F. The Effect of Formalin on Botulinum Toxins A, B and C. J. Immunol. 1929, 16, 391–401. [Google Scholar] [CrossRef]
- Aquino, M.R.; Bingemann, T.A.; Nanda, A.; Maples, K.M. Delayed allergic skin reactions to vaccines. Allergy Asthma Proc. 2022, 43, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Dewitte, H. The dawn of mRNA vaccines: The COVID-19 case. J. Control. Release 2021, 333, 511–520. [Google Scholar] [CrossRef]
- Jain, S.; Venkataraman, A.; Wechsler, M.E.; Peppas, N.A. Messenger RNA-based vaccines: Past, present, and future directions in the context of the COVID-19 pandemic. Adv. Drug. Deliv. Rev. 2021, 179, 114000. [Google Scholar] [CrossRef]
- Delany, I.; Rappuoli, R.; De Gregorio, E. Vaccines for the 21st century. EMBO Mol. Med. 2014, 6, 708–720. [Google Scholar] [CrossRef]
- Akkaya, M.; Kwak, K.; Pierce, S.K. B cell memory: Building two walls of protection against pathogens. Nat. Rev. Immunol. 2020, 20, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Minor, P.D. Live attenuated vaccines: Historical successes and current challenges. Virology 2015, 479–480, 379–392. [Google Scholar] [CrossRef]
- Strebel, P.M.; Papania, M.J.; Fiebelkorn, A.P.; Halsey, N.A. 20—Measles vaccine. In Vaccines, 6th ed.; Plotkin, S.A., Orenstein, W.A., Offit, P.A., Eds.; W.B. Saunders: London, UK, 2013; pp. 352–387. [Google Scholar] [CrossRef]
- Lieberman, J.M.; Williams, W.R.; Miller, J.M.; Black, S.; Shinefield, H.; Henderson, F.; Marchant, C.D.; Werzberger, A.; Halperin, S.; Hartzel, J.; et al. The safety and immunogenicity of a quadrivalent measles, mumps, rubella and varicella vaccine in healthy children: A study of manufacturing consistency and persistence of antibody. Pediatr. Infect. Dis. J. 2006, 25, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Patja, A.; Davidkin, I.; Kurki, T.; Kallio, M.J.; Valle, M.; Peltola, H. Serious adverse events after measles-mumps-rubella vaccination during a fourteen-year prospective follow-up. Pediatr. Infect. Dis. J. 2000, 19, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Pace, J.L.; Rossi, H.A.; Esposito, V.M.; Frey, S.M.; Tucker, K.D.; Walker, R.I. Inactivated whole-cell bacterial vaccines: Current status and novel strategies. Vaccine 1998, 16, 1563–1574. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.S.; Garon, J.; Seib, K.; Orenstein, W.A. Polio vaccination: Past, present and future. Future Microbiol. 2015, 10, 791–808. [Google Scholar] [CrossRef]
- Hird, T.R.; Grassly, N.C. Systematic review of mucosal immunity induced by oral and inactivated poliovirus vaccines against virus shedding following oral poliovirus challenge. PLoS Pathog. 2012, 8, e1002599. [Google Scholar] [CrossRef]
- World Health Organization. Supplementary Information on Vaccine Safety. Part 2: Background Rates of Adverse Events Following Immunization. World Health Organization. 2000. Available online: https://apps.who.int/iris/handle/10665/66675 (accessed on 1 July 2023).
- Ren, Z.; Sun, R.; Cui, G.; Wang, H.; Zhang, D.; Li, J.; Zhang, Y.; Yu, Z. Effects of Inactivated Vaccination on Humoral Immune Responses in Patients Infected with Delta or Omicron Variants. J. Infect. Dis. 2022, 226, 1120–1122. [Google Scholar] [CrossRef]
- Excler, J.L.; Saville, M.; Berkley, S.; Kim, J.H. Vaccine development for emerging infectious diseases. Nat. Med. 2021, 27, 591–600. [Google Scholar] [CrossRef]
- CDC. Adverse Reaction. 2022. Available online: https://www.cdc.gov/rabies/specific_groups/hcp/adverse_reaction.html#:~:text=Immediate%20hypersensitivity%20reactions%20have%20been,6%2D14%20days%20after%20boosters (accessed on 1 July 2023).
- Liang, J.L.; Tiwari, T.; Moro, P.; Messonnier, N.E.; Reingold, A.; Sawyer, M.; Clark, T.A. Prevention of Pertussis, Tetanus, and Diphtheria with Vaccines in the United States: Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm. Rep. 2018, 67, 1–44. [Google Scholar] [CrossRef]
- McQuillan, G.M.; Kruszon-Moran, D.; Deforest, A.; Chu, S.Y.; Wharton, M. Serologic immunity to diphtheria and tetanus in the United States. Ann. Intern. Med. 2002, 136, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Angsantikul, P.; Fang, R.H.; Zhang, L. Toxoid Vaccination against Bacterial Infection Using Cell Membrane-Coated Nanoparticles. Bioconjug. Chem. 2018, 29, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Shaw, C.A.; Starnbach, M.N. Stimulation of CD8 + T cells following diphtheria toxin-mediated antigen delivery into dendritic cells. Infect. Immun. 2006, 74, 1001–1008. [Google Scholar] [CrossRef]
- Skibinski, D.A.; Baudner, B.C.; Singh, M.; O’Hagan, D.T. Combination vaccines. J. Glob. Infect. Dis. 2011, 3, 63–72. [Google Scholar] [CrossRef]
- Farrar, J.; Newton, C. Neurological aspects of tropical disease. J. Neurol. Neurosurg. Psychiatry 2000, 68, 135–136. [Google Scholar] [CrossRef] [PubMed]
- Stock, I. Tetanus and Clostridium tetani—A brief review. Med. Monatsschr. Pharm. 2015, 38, 57–60. [Google Scholar]
- Rhinesmith, E.; Fu, L. Tetanus Disease, Treatment, Management. Pediatr. Rev. 2018, 39, 430–432. [Google Scholar] [CrossRef]
- Goretzki, K.; Habermann, E. Enzymatic hydrolysis of tetanus toxin by intrinsic and extrinsic proteases. Characterization of the fragments by monoclonal antibodies. Med. Microbiol. Immunol. 1985, 174, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Farrar, J.J.; Yen, L.M.; Cook, T.; Fairweather, N.; Binh, N.; Parry, J.; Parry, C.M. Tetanus. J. Neurol. Neurosurg. Psychiatry 2000, 69, 292–301. [Google Scholar] [CrossRef]
- Behrensdorf-Nicol, H.A.; Kegel, B.; Bonifas, U.; Silberbach, K.; Klimek, J.; Weiber, K.; Kramer, B. Residual enzymatic activity of the tetanus toxin light chain present in tetanus toxoid batches used for vaccine production. Vaccine 2008, 26, 3835–3841. [Google Scholar] [CrossRef]
- Stojicevic, I.; Dimitrijevic, L.; Dovezenski, N.; Zivkovic, I.; Petrusic, V.; Marinkovic, E.; Inic-Kanada, A.; Stojanovic, M. Tetanus toxoid purification: Chromatographic procedures as an alternative to ammonium-sulphate precipitation. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 2213–2219. [Google Scholar] [CrossRef]
- Rossetto, O.; Montecucco, C. Tables of Toxicity of Botulinum and Tetanus Neurotoxins. Toxins 2019, 11, 686. [Google Scholar] [CrossRef] [PubMed]
- Thaysen-Andersen, M.; Jorgensen, S.B.; Wilhelmsen, E.S.; Petersen, J.W.; Hojrup, P. Investigation of the detoxification mechanism of formaldehyde-treated tetanus toxin. Vaccine 2007, 25, 2213–2227. [Google Scholar] [CrossRef] [PubMed]
- Michiels, T.J.M.; Meiring, H.D.; Jiskoot, W.; Kersten, G.F.A.; Metz, B. Formaldehyde treatment of proteins enhances proteolytic degradation by the endo-lysosomal protease cathepsin S. Sci. Rep. 2020, 10, 11535. [Google Scholar] [CrossRef]
- Fan, Y.C.; Chiu, H.C.; Chen, L.K.; Chang, G.J.; Chiou, S.S. Formalin Inactivation of Japanese Encephalitis Virus Vaccine Alters the Antigenicity and Immunogenicity of a Neutralization Epitope in Envelope Protein Domain III. PLoS Neglected Trop. Dis. 2015, 9, e0004167. [Google Scholar] [CrossRef]
- Metz, B.; Michiels, T.; Uittenbogaard, J.; Danial, M.; Tilstra, W.; Meiring, H.D.; Hennink, W.E.; Crommelin, D.J.A.; Kersten, G.F.A.; Jiskoot, W. Identification of Formaldehyde-Induced Modifications in Diphtheria Toxin. J. Pharm. Sci. 2020, 109, 543–557. [Google Scholar] [CrossRef]
- Michiels, T.J.M.; Schoneich, C.; Hamzink, M.R.J.; Meiring, H.D.; Kersten, G.F.A.; Jiskoot, W.; Metz, B. Novel Formaldehyde-Induced Modifications of Lysine Residue Pairs in Peptides and Proteins: Identification and Relevance to Vaccine Development. Mol. Pharm. 2020, 17, 4375–4385. [Google Scholar] [CrossRef]
- Mc, C.J.; Trafton, M.Z. Immune responses and reactions to diphtheria and tetanus toxoids, with pertussis vaccine, aluminum phosphate precipitated. N. Engl. J. Med. 1950, 243, 442–444. [Google Scholar] [CrossRef]
- Ghotloo, S.; Golsaz-Shirazi, F.; Amiri, M.M.; Jeddi-Tehrani, M.; Shokri, F. Epitope Mapping of Tetanus Toxin by Monoclonal Antibodies: Implication for Immunotherapy and Vaccine Design. Neurotox. Res. 2020, 37, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Blencowe, H.; Lawn, J.; Vandelaer, J.; Roper, M.; Cousens, S. Tetanus toxoid immunization to reduce mortality from neonatal tetanus. Int. J. Epidemiol. 2010, 39 (Suppl. S1), i102–i109. [Google Scholar] [CrossRef]
- Kyu, H.H.; Mumford, J.E.; Stanaway, J.D.; Barber, R.M.; Hancock, J.R.; Vos, T.; Murray, C.J.; Naghavi, M. Mortality from tetanus between 1990 and 2015: Findings from the global burden of disease study 2015. BMC Public Health 2017, 17, 179. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.C.; Efstratiou, A.; Mokrousov, I.; Mutreja, A.; Das, B.; Ramamurthy, T. Diphtheria. Nat. Rev. Dis. Prim. 2019, 5, 81. [Google Scholar] [CrossRef]
- Holbourn, K.P.; Shone, C.C.; Acharya, K.R. A family of killer toxins. Exploring the mechanism of ADP-ribosylating toxins. FEBS J. 2006, 273, 4579–4593. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Diptheria Reported Cases by WHO Region. In WHO Site; World Health Organization: Geneva, Switzerland, 2023. [Google Scholar]
- Gill, D.M. Bacterial toxins: A table of lethal amounts. Microbiol. Rev. 1982, 46, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.K.; Smith, T.J.; Helma, C.H.; Ticknor, L.O.; Foley, B.T.; Svensson, R.T.; Brown, J.L.; Johnson, E.A.; Smith, L.A.; Okinaka, R.T.; et al. Genetic diversity among Botulinum Neurotoxin-producing clostridial strains. J. Bacteriol. 2007, 189, 818–832. [Google Scholar] [CrossRef]
- Schiavo, G.; Matteoli, M.; Montecucco, C. Neurotoxins affecting neuroexocytosis. Physiol. Rev. 2000, 80, 717–766. [Google Scholar] [CrossRef]
- Montecucco, C.; Rossetto, O.; Schiavo, G. Presynaptic receptor arrays for clostridial neurotoxins. Trends Microbiol. 2004, 12, 442–446. [Google Scholar] [CrossRef]
- Lam, K.H.; Yao, G.; Jin, R. Diverse binding modes, same goal: The receptor recognition mechanism of botulinum neurotoxin. Prog. Biophys. Mol. Biol. 2015, 117, 225–231. [Google Scholar] [CrossRef]
- Lacy, D.B.; Tepp, W.; Cohen, A.C.; DasGupta, B.R.; Stevens, R.C. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat. Struct. Biol. 1998, 5, 898–902. [Google Scholar] [CrossRef]
- Whitemarsh, R.C.; Tepp, W.H.; Johnson, E.A.; Pellett, S. Persistence of botulinum neurotoxin a subtypes 1–5 in primary rat spinal cord cells. PLoS ONE 2014, 9, e90252. [Google Scholar] [CrossRef]
- Pellett, S.; Bradshaw, M.; Tepp, W.H.; Pier, C.L.; Whitemarsh, R.C.M.; Chen, C.; Barbieri, J.T.; Johnson, E.A. The Light Chain Defines the Duration of Action of Botulinum Toxin Serotype A Subtypes. mBio 2018, 9, 00089-18. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.P.; Barbieri, J.T. Light Chain Diversity among the Botulinum Neurotoxins. Toxins 2018, 10, 268. [Google Scholar] [CrossRef] [PubMed]
- Hatheway, C.L. Botulism: The present status of the disease. Curr. Top. Microbiol. Immunol. 1995, 195, 55–75. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Marquez, M.A. Botulism. Pediatr. Rev. 2016, 37, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Sundeen, G.; Barbieri, J.T. Vaccines against Botulism. Toxins 2017, 9, 268. [Google Scholar] [CrossRef]
- Karalewitz, A.P.; Barbieri, J.T. Vaccines against botulism. Curr. Opin. Microbiol. 2012, 15, 317–324. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Notice of CDC’s discontinuation of investigational pentavalent (ABCDE) botulinum toxoid vaccine for workers at risk for occupational exposure to botulinum toxins. MMWR Morb. Mortal. Wkly. Rep. 2011, 60, 1454–1455. [Google Scholar]
- Rusnak, J.M.; Smith, L.A. Botulinum neurotoxin vaccines: Past history and recent developments. Hum. Vaccin. 2009, 5, 794–805. [Google Scholar] [CrossRef]
- Smith, L.A.; Rusnak, J.M. Botulinum neurotoxin vaccines: Past, present, and future. Crit. Rev. Immunol. 2007, 27, 303–318. [Google Scholar] [CrossRef]
- Pier, C.L.; Tepp, W.H.; Bradshaw, M.; Johnson, E.A.; Barbieri, J.T.; Baldwin, M.R. Recombinant holotoxoid vaccine against botulism. Infect. Immun. 2008, 76, 437–442. [Google Scholar] [CrossRef]
- Bradshaw, M.; Goodnough, M.C.; Johnson, E.A. Conjugative transfer of the Escherichia coli-Clostridium perfringens shuttle vector pJIR1457 to Clostridium botulinum type A strains. Plasmid 1998, 40, 233–237. [Google Scholar] [CrossRef]
- Johnson, E.A. Clostridial toxins as therapeutic agents: Benefits of nature’s most toxic proteins. Annu. Rev. Microbiol. 1999, 53, 551–575. [Google Scholar] [CrossRef] [PubMed]
- Whitemarsh, R.C.; Tepp, W.H.; Bradshaw, M.; Lin, G.; Pier, C.L.; Scherf, J.M.; Johnson, E.A.; Pellett, S. Characterization of botulinum neurotoxin A subtypes 1 through 5 by investigation of activities in mice, in neuronal cell cultures, and in vitro. Infect. Immun. 2013, 81, 3894–3902. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.A. Development of recombinant vaccines for botulinum neurotoxin. Toxicon 1998, 36, 1539–1548. [Google Scholar] [CrossRef]
- Torii, Y.; Sugimoto, N.; Kohda, T.; Kozaki, S.; Morokuma, K.; Horikawa, Y.; Ginnaga, A.; Yamamoto, A.; Takahashi, M. Clinical Study of New Tetravalent (Type A, B, E, and F) Botulinum Toxoid Vaccine Derived from M Toxin in Japan. Jpn. J. Infect. Dis. 2017, 70, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, R.; Kohda, T.; Kataoka, K.; Ihara, H.; Kozaki, S.; Pascual, D.W.; Staats, H.F.; Kiyono, H.; McGhee, J.R.; Fujihashi, K. A novel neurotoxoid vaccine prevents mucosal botulism. J. Immunol. 2005, 174, 2190–2195. [Google Scholar] [CrossRef]
- Torii, Y.; Tokumaru, Y.; Kawaguchi, S.; Izumi, N.; Maruyama, S.; Mukamoto, M.; Kozaki, S.; Takahashi, M. Production and immunogenic efficacy of botulinum tetravalent (A, B, E, F) toxoid. Vaccine 2002, 20, 2556–2561. [Google Scholar] [CrossRef]
- Dertzbaugh, M.T. Genetically engineered vaccines: An overview. Plasmid 1998, 39, 100–113. [Google Scholar] [CrossRef]
- Greenberg, L. A New Approach to Bacterial Vaccines. Can. Med. Assoc. J. 1963, 89, 396–402. [Google Scholar]
- Sivakumar, S.M.; Safhi, M.M.; Kannadasan, M.; Sukumaran, N. Vaccine adjuvants—Current status and prospects on controlled release adjuvancity. Saudi. Pharm. J. 2011, 19, 197–206. [Google Scholar] [CrossRef]
- Gupta, R.K.; Rost, B.E.; Relyveld, E.; Siber, G.R. Adjuvant properties of aluminum and calcium compounds. Pharm. Biotechnol. 1995, 6, 229–248. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Dinamarca, D.A.; Salazar, M.L.; Castillo, B.N.; Manubens, A.; Vasquez, A.E.; Salazar, F.; Becker, M.I. Protein-Based Adjuvants for Vaccines as Immunomodulators of the Innate and Adaptive Immune Response: Current Knowledge, Challenges, and Future Opportunities. Pharmaceutics 2022, 14, 1671. [Google Scholar] [CrossRef] [PubMed]
- Pulendran, B.; Arunachalam, P.S.; O’Hagan, D.T. Emerging concepts in the science of vaccine adjuvants. Nat. Rev. Drug Discov. 2021, 20, 454–475. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.K.; Mahajan, P.; Singh, N.K.; Gupta, A.; Aggarwal, R.; Rappuoli, R.; Johri, A.K. New-age vaccine adjuvants, their development, and future perspective. Front. Immunol. 2023, 14, 1043109. [Google Scholar] [CrossRef]
- Slifka, M.K.; Amanna, I. How advances in immunology provide insight into improving vaccine efficacy. Vaccine 2014, 32, 2948–2957. [Google Scholar] [CrossRef]
- Coffman, R.L.; Sher, A.; Seder, R.A. Vaccine adjuvants: Putting innate immunity to work. Immunity 2010, 33, 492–503. [Google Scholar] [CrossRef]
- Schwarz, T.F.; Horacek, T.; Knuf, M.; Damman, H.G.; Roman, F.; Drame, M.; Gillard, P.; Jilg, W. Single dose vaccination with AS03-adjuvanted H5N1 vaccines in a randomized trial induces strong and broad immune responsiveness to booster vaccination in adults. Vaccine 2009, 27, 6284–6290. [Google Scholar] [CrossRef]
- Zhao, H.; Zhou, X.; Zhou, Y.H. Hepatitis B vaccine development and implementation. Hum. Vaccine Immunother. 2020, 16, 1533–1544. [Google Scholar] [CrossRef]
- Adkins, J.C.; Wagstaff, A.J. Recombinant hepatitis B vaccine: A review of its immunogenicity and protective efficacy against hepatitis B. BioDrugs 1998, 10, 137–158. [Google Scholar] [CrossRef]
- Govan, V.A. A novel vaccine for cervical cancer: Quadrivalent human papillomavirus (types 6, 11, 16 and 18) recombinant vaccine (Gardasil). Ther. Clin. Risk Manag. 2008, 4, 65–70. [Google Scholar] [CrossRef]
- Shinefield, H.R. Overview of the development and current use of CRM(197) conjugate vaccines for pediatric use. Vaccine 2010, 28, 4335–4339. [Google Scholar] [CrossRef] [PubMed]
- Rappuoli, R. Isolation and characterization of Corynebacterium diphtheriae nontandem double lysogens hyperproducing CRM197. Appl. Environ. Microbiol. 1983, 46, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Ghani, K.; Caruso, M. Diphtheria toxin mutant CRM197 is an inhibitor of protein synthesis that induces cellular toxicity. Toxicon 2008, 51, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, T.; Ohishi, M.; Miyamoto, S.; Mizushima, H.; Iwamoto, R.; Mekada, E. Diphtheria toxin mutant CRM197 possesses weak EF2-ADP-ribosyl activity that potentiates its anti-tumorigenic activity. J. Biochem. 2007, 142, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Rabadi, T.; Brady, M.F. Tetanus Toxoid. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- World Health Organization. Vaccine Safety Basics; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Liu, F.J.; Shi, D.Y.; Li, Z.Y.; Lu, J.S.; Wang, R.; Pang, X.B.; Yang, Z.X.; Yu, Y.Z. Evaluation of a recombinant tetanus toxin subunit vaccine. Toxicon 2020, 187, 75–81. [Google Scholar] [CrossRef]
- Chang, M.J.; Ollivault-Shiflett, M.; Schuman, R.; Ngoc Nguyen, S.; Kaltashov, I.A.; Bobst, C.; Rajagopal, S.P.; Przedpelski, A.; Barbieri, J.T.; Lees, A. Genetically detoxified tetanus toxin as a vaccine and conjugate carrier protein. Vaccine 2022, 40, 5103–5113. [Google Scholar] [CrossRef]
- Rasetti-Escargueil, C.; Popoff, M.R. Antibodies and Vaccines against Botulinum Toxins: Available Measures and Novel Approaches. Toxins 2019, 11, 528. [Google Scholar] [CrossRef]
- LaPenotiere, H.F.; Clayton, M.A.; Middlebrook, J.L. Expression of a large, nontoxic fragment of botulinum neurotoxin serotype A and its use as an immunogen. Toxicon 1995, 33, 1383–1386. [Google Scholar] [CrossRef]
- Webb, R.P.; Smith, L.A. What next for botulism vaccine development? Expert Rev. Vaccines 2013, 12, 481–492. [Google Scholar] [CrossRef]
- Baldwin, M.R.; Tepp, W.H.; Przedpelski, A.; Pier, C.L.; Bradshaw, M.; Johnson, E.A.; Barbieri, J.T. Subunit vaccine against the seven serotypes of botulism. Infect. Immun. 2008, 76, 1314–1318. [Google Scholar] [CrossRef]
- Przedpelski, A.; Tepp, W.H.; Kroken, A.R.; Fu, Z.; Kim, J.J.; Johnson, E.A.; Barbieri, J.T. Enhancing the protective immune response against botulism. Infect. Immun. 2013, 81, 2638–2644. [Google Scholar] [CrossRef]
- Baldwin, M.R.; Tepp, W.H.; Pier, C.L.; Bradshaw, M.; Ho, M.; Wilson, B.A.; Fritz, R.B.; Johnson, E.A.; Barbieri, J.T. Characterization of the antibody response to the receptor binding domain of botulinum neurotoxin serotypes A and E. Infect. Immun. 2005, 73, 6998–7005. [Google Scholar] [CrossRef] [PubMed]
- Khouri, J.M.; Motter, R.N.; Arnon, S.S. Safety and immunogenicity of investigational recombinant botulinum vaccine, rBV A/B, in volunteers with pre-existing botulinum toxoid immunity. Vaccine 2018, 36, 2041–2048. [Google Scholar] [CrossRef] [PubMed]
- Webb, R.P.; Smith, T.J.; Smith, L.A.; Wright, P.M.; Guernieri, R.L.; Brown, J.L.; Skerry, J.C. Recombinant Botulinum Neurotoxin Hc Subunit (BoNT Hc) and Catalytically Inactive Clostridium botulinum Holoproteins (ciBoNT HPs) as Vaccine Candidates for the Prevention of Botulism. Toxins 2017, 9, 269. [Google Scholar] [CrossRef] [PubMed]
- Byrne, M.P.; Smith, L.A. Development of vaccines for prevention of botulism. Biochimie 2000, 82, 955–966. [Google Scholar] [CrossRef]
- Band, P.A.; Blais, S.; Neubert, T.A.; Cardozo, T.J.; Ichtchenko, K. Recombinant derivatives of botulinum neurotoxin A engineered for trafficking studies and neuronal delivery. Protein Expr. Purif. 2010, 71, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Tepp, W.H.; Bradshaw, M.; Gardner, A.P.; Kaufman, R.L.; Barbieri, J.T.; Pellett, S. Botulinum Neurotoxin A4 Has a 1000-Fold Reduced Potency Due to Three Single Amino Acid Alterations in the Protein Receptor Binding Domain. Int. J. Mol. Sci. 2023, 24, 5690. [Google Scholar] [CrossRef]
- Bradshaw, M.; Tepp, W.H.; Whitemarsh, R.C.; Pellett, S.; Johnson, E.A. Holotoxin Activity of Botulinum Neurotoxin Subtype A4 Originating from a Nontoxigenic Clostridium botulinum Expression System. Appl. Environ. Microbiol. 2014, 80, 7415–7422. [Google Scholar] [CrossRef]
- Dolly, J.O.; Wang, J.; Zurawski, T.H.; Meng, J. Novel therapeutics based on recombinant botulinum neurotoxins to normalize the release of transmitters and pain mediators. FEBS J. 2011, 278, 4454–4466. [Google Scholar] [CrossRef]
- Rummel, A.; Mahrhold, S.; Bigalke, H.; Binz, T. Exchange of the H(CC) domain mediating double receptor recognition improves the pharmacodynamic properties of botulinum neurotoxin. FEBS J. 2011, 278, 4506–4515. [Google Scholar] [CrossRef]
- Webb, R.P.; Smith, T.J.; Wright, P.; Brown, J.; Smith, L.A. Production of catalytically inactive BoNT/A1 holoprotein and comparison with BoNT/A1 subunit vaccines against toxin subtypes A1, A2, and A3. Vaccine 2009, 27, 4490–4497. [Google Scholar] [CrossRef]
- Webb, R.P. Engineering of Botulinum Neurotoxins for Biomedical Applications. Toxins 2018, 10, 231. [Google Scholar] [CrossRef]
- Smith, T.J.; Lou, J.; Geren, I.N.; Forsyth, C.M.; Tsai, R.; Laporte, S.L.; Tepp, W.H.; Bradshaw, M.; Johnson, E.A.; Smith, L.A.; et al. Sequence variation within botulinum neurotoxin serotypes impacts antibody binding and neutralization. Infect. Immun. 2005, 73, 5450–5457. [Google Scholar] [CrossRef]
- Przedpelski, A.; Tepp, W.H.; Zuverink, M.; Johnson, E.A.; Pellet, S.; Barbieri, J.T. Enhancing toxin-based vaccines against botulism. Vaccine 2018, 36, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lu, J.; Tan, X.; Wang, R.; Xu, Q.; Yu, Y.; Yang, Z. Functional EL-HN Fragment as a Potent Candidate Vaccine for the Prevention of Botulinum Neurotoxin Serotype E. Toxins 2022, 14, 135. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, B.; Lu, J.; Liu, X.; Tan, X.; Wang, R.; Du, P.; Yu, S.; Xu, Q.; Pang, X.; et al. Biological and Immunological Characterization of a Functional L-HN Derivative of Botulinum Neurotoxin Serotype F. Toxins 2023, 15, 200. [Google Scholar] [CrossRef]
- Ayyar, B.V.; Tajhya, R.B.; Beeton, C.; Atassi, M.Z. Antigenic sites on the HN domain of botulinum neurotoxin A stimulate protective antibody responses against active toxin. Sci. Rep. 2015, 5, 15776. [Google Scholar] [CrossRef] [PubMed]
- Atassi, M.Z.; Dolimbek, B.Z. Mapping of the antibody-binding regions on the HN-domain (residues 449-859) of botulinum neurotoxin A with antitoxin antibodies from four host species. Full profile of the continuous antigenic regions of the H-chain of botulinum neurotoxin A. Protein J. 2004, 23, 39–52. [Google Scholar] [CrossRef]
- Atassi, M.Z. Molecular basis of immunogenicity to botulinum neurotoxins and uses of the defined antigenic regions. Toxicon 2015, 107 Pt A, 50–58. [Google Scholar] [CrossRef]
- Shone, C.; Agostini, H.; Clancy, J.; Gu, M.; Yang, H.H.; Chu, Y.; Johnson, V.; Taal, M.; McGlashan, J.; Brehm, J.; et al. Bivalent recombinant vaccine for botulinum neurotoxin types A and B based on a polypeptide comprising their effector and translocation domains that is protective against the predominant A and B subtypes. Infect. Immun. 2009, 77, 2795–2801. [Google Scholar] [CrossRef]
- Binz, T.; Bade, S.; Rummel, A.; Kollewe, A.; Alves, J. Arg(362) and Tyr(365) of the botulinum neurotoxin type a light chain are involved in transition state stabilization. Biochemistry 2002, 41, 1717–1723. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Binz, T.; Swaminathan, S. Analysis of active site residues of botulinum neurotoxin E by mutational, functional, and structural studies: Glu335Gln is an apoenzyme. Biochemistry 2005, 44, 8291–8302. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, E.; Janardhanan, P.; Patel, K.; Riding, S.; Cai, S.; Singh, B.R. In Vivo Toxicity and Immunological Characterization of Detoxified Recombinant Botulinum Neurotoxin Type A. Pharm. Res. 2016, 33, 639–652. [Google Scholar] [CrossRef]
- Gu, S.; Rumpel, S.; Zhou, J.; Strotmeier, J.; Bigalke, H.; Perry, K.; Shoemaker, C.B.; Rummel, A.; Jin, R. Botulinum neurotoxin is shielded by NTNHA in an interlocked complex. Science 2012, 335, 977–981. [Google Scholar] [CrossRef]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Brian, D.A.; Baric, R.S. Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 2005, 287, 1–30. [Google Scholar] [CrossRef]
- Chilamakuri, R.; Agarwal, S. COVID-19: Characteristics and Therapeutics. Cells 2021, 10, 206. [Google Scholar] [CrossRef]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Hadj Hassine, I. COVID-19 vaccines and variants of concern: A review. Rev. Med. Virol. 2022, 32, e2313. [Google Scholar] [CrossRef]
- Tregoning, J.S.; Flight, K.E.; Higham, S.L.; Wang, Z.; Pierce, B.F. Progress of the COVID-19 vaccine effort: Viruses, vaccines and variants versus efficacy, effectiveness and escape. Nat. Rev. Immunol. 2021, 21, 626–636. [Google Scholar] [CrossRef]
- Sharif, N.; Alzahrani, K.J.; Ahmed, S.N.; Dey, S.K. Efficacy, Immunogenicity and Safety of COVID-19 Vaccines: A Systematic Review and Meta-Analysis. Front. Immunol. 2021, 12, 714170. [Google Scholar] [CrossRef]
- Pollet, J.; Chen, W.H.; Strych, U. Recombinant protein vaccines, a proven approach against coronavirus pandemics. Adv. Drug. Deliv. Rev. 2021, 170, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Maurya, V.K.; Prasad, A.K.; Bhatt, M.L.B.; Saxena, S.K. Structural, glycosylation and antigenic variation between 2019 novel coronavirus (2019-nCoV) and SARS coronavirus (SARS-CoV). Virusdisease 2020, 31, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Drosten, C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antivir. Res. 2014, 101, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Enose-Akahata, Y.; Wang, L.; Almsned, F.; Johnson, K.R.; Mina, Y.; Ohayon, J.; Wang, X.W.; Jacobson, S. The repertoire of CSF antiviral antibodies in patients with neuroinflammatory diseases. Sci. Adv. 2023, 9, eabq6978. [Google Scholar] [CrossRef]
- Elko, E.A.; Nelson, G.A.; Mead, H.L.; Kelley, E.J.; Carvalho, S.T.; Sarbo, N.G.; Harms, C.E.; Le Verche, V.; Cardoso, A.A.; Ely, J.L.; et al. COVID-19 vaccination elicits an evolving, cross-reactive antibody response to epitopes conserved with endemic coronavirus spike proteins. Cell Rep. 2022, 40, 111022. [Google Scholar] [CrossRef]
- Xiaojie, S.; Yu, L.; Lei, Y.; Guang, Y.; Min, Q. Neutralizing antibodies targeting SARS-CoV-2 spike protein. Stem Cell Res. 2020, 50, 102125. [Google Scholar] [CrossRef]
- Novavax. Novavax’s Nuvaxovid™ Receives Positive CHMP Opinion for Full Marketing Authorization for the Prevention of COVID in the EU. 2023. Available online: https://ir.novavax.com/press-releases/2023-05-26-Novavaxs-Nuvaxovid-TM-Receives-Positive-CHMP-Opinion-for-Full-Marketing-Authorization-for-the-Prevention-of-COVID-in-the-EU (accessed on 1 July 2023).
- Heath, P.T.; Galiza, E.P.; Baxter, D.N.; Boffito, M.; Browne, D.; Burns, F.; Chadwick, D.R.; Clark, R.; Cosgrove, C.; Galloway, J.; et al. Safety and Efficacy of NVX-CoV2373 COVID-19 Vaccine. N. Engl. J. Med. 2021, 385, 1172–1183. [Google Scholar] [CrossRef]
- Dunkle, L.M.; Kotloff, K.L.; Gay, C.L.; Anez, G.; Adelglass, J.M.; Barrat Hernandez, A.Q.; Harper, W.L.; Duncanson, D.M.; McArthur, M.A.; Florescu, D.F.; et al. Efficacy and Safety of NVX-CoV2373 in Adults in the United States and Mexico. N. Engl. J. Med. 2022, 386, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Toback, S.; Galiza, E.; Cosgrove, C.; Galloway, J.; Goodman, A.L.; Swift, P.A.; Rajaram, S.; Graves-Jones, A.; Edelman, J.; Burns, F.; et al. Safety, immunogenicity, and efficacy of a COVID-19 vaccine (NVX-CoV2373) co-administered with seasonal influenza vaccines: An exploratory substudy of a randomised, observer-blinded, placebo-controlled, phase 3 trial. Lancet Respir. Med. 2022, 10, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Trougakos, I.P.; Terpos, E.; Alexopoulos, H.; Politou, M.; Paraskevis, D.; Scorilas, A.; Kastritis, E.; Andreakos, E.; Dimopoulos, M.A. Adverse effects of COVID-19 mRNA vaccines: The spike hypothesis. Trends Mol. Med. 2022, 28, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.; Xu, Q. Current Developments and Challenges of mRNA Vaccines. Annu. Rev. Biomed. Eng. 2022, 24, 85–109. [Google Scholar] [CrossRef]
- Rosa, S.S.; Prazeres, D.M.F.; Azevedo, A.M.; Marques, M.P.C. mRNA vaccines manufacturing: Challenges and bottlenecks. Vaccine 2021, 39, 2190–2200. [Google Scholar] [CrossRef]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K.J. Developing mRNA-vaccine technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef]
- Teo, S.P. Review of COVID-19 mRNA Vaccines: BNT162b2 and mRNA-1273. J. Pharm. Pract. 2022, 35, 947–951. [Google Scholar] [CrossRef]
- Wang, L.; Davis, P.B.; Kaelber, D.C.; Volkow, N.D.; Xu, R. Comparison of mRNA-1273 and BNT162b2 Vaccines on Breakthrough SARS-CoV-2 Infections, Hospitalizations, and Death during the Delta-Predominant Period. JAMA 2022, 327, 678–680. [Google Scholar] [CrossRef]
- Garreffa, E.; Hamad, A.; O’Sullivan, C.C.; Hazim, A.Z.; York, J.; Puri, S.; Turnbull, A.; Robertson, J.F.; Goetz, M.P. Regional lymphadenopathy following COVID-19 vaccination: Literature review and considerations for patient management in breast cancer care. Eur. J. Cancer 2021, 159, 38–51. [Google Scholar] [CrossRef]
- Shimabukuro, T. Allergic reactions including anaphylaxis after receipt of the first dose of Moderna COVID-19 vaccine—United States, 21 December 2020–10 January 2021. Am. J. Transpl. 2021, 21, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- CDC COVID-19 Response Team; Food and Drug Administration. Allergic Reactions Including Anaphylaxis After Receipt of the First Dose of Moderna COVID-19 Vaccine—United States, 21 December 2020–10 January 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 125–129. [Google Scholar] [CrossRef]
- Buchan, S.A.; Alley, S.; Seo, C.Y.; Johnson, C.; Kwong, J.C.; Nasreen, S.; Thampi, N.; Lu, D.; Harris, T.M.; Calzavara, A.; et al. Myocarditis or Pericarditis Events After BNT162b2 Vaccination in Individuals Aged 12 to 17 Years in Ontario, Canada. JAMA Pediatr. 2023, 177, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.E.; Taub, I.B.; Kaelber, D.C. Risk of Myocarditis from COVID-19 Infection in People Under Age 20: A Population-Based Analysis. medRxiv 2022. medRxiv:2021.07.23.21260998. [Google Scholar] [CrossRef]
- Ewer, K.; Sebastian, S.; Spencer, A.J.; Gilbert, S.; Hill, A.V.S.; Lambe, T. Chimpanzee adenoviral vectors as vaccines for outbreak pathogens. Hum. Vaccin. Immunother. 2017, 13, 3020–3032. [Google Scholar] [CrossRef]
- Custers, J.; Kim, D.; Leyssen, M.; Gurwith, M.; Tomaka, F.; Robertson, J.; Heijnen, E.; Condit, R.; Shukarev, G.; Heerwegh, D.; et al. Vaccines based on replication incompetent Ad26 viral vectors: Standardized template with key considerations for a risk/benefit assessment. Vaccine 2021, 39, 3081–3101. [Google Scholar] [CrossRef]
- Koger-Pease, C.; Perera, D.J.; Ndao, M. Recent Advances in the Development of Adenovirus-Vectored Vaccines for Parasitic Infections. Pharmaceuticals 2023, 16, 334. [Google Scholar] [CrossRef]
- Francis, A.I.; Ghany, S.; Gilkes, T.; Umakanthan, S. Review of COVID-19 vaccine subtypes, efficacy and geographical distributions. Postgrad. Med. J. 2022, 98, 389–394. [Google Scholar] [CrossRef]
- Food and Drug Administration (USFDA). Coronavirus (COVID-19) Update: FDA Limits Use of Janssen COVID-19 Vaccine to Certain Individuals. 2022. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-limits-use-janssen-covid-19-vaccine-certain-individuals (accessed on 1 July 2023).
- Bozkurt, B. Shedding Light on Mechanisms of Myocarditis with COVID-19 mRNA Vaccines. Circulation 2023, 147, 877–880. [Google Scholar] [CrossRef]
- Hajjo, R.; Sabbah, D.A.; Bardaweel, S.K.; Tropsha, A. Shedding the Light on Post-Vaccine Myocarditis and Pericarditis in COVID-19 and Non-COVID-19 Vaccine Recipients. Vaccines 2021, 9, 1186. [Google Scholar] [CrossRef]
- Thran, M.; Mukherjee, J.; Ponisch, M.; Fiedler, K.; Thess, A.; Mui, B.L.; Hope, M.J.; Tam, Y.K.; Horscroft, N.; Heidenreich, R.; et al. mRNA mediates passive vaccination against infectious agents, toxins, and tumors. EMBO Mol. Med. 2017, 9, 1434–1447. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, J.; Ondeck, C.A.; Tremblay, J.M.; Archer, J.; Debatis, M.; Foss, A.; Awata, J.; Erasmus, J.H.; McNutt, P.M.; Shoemaker, C.B. Intramuscular delivery of formulated RNA encoding six linked nanobodies is highly protective for exposures to three Botulinum neurotoxin serotypes. Sci. Rep. 2022, 12, 11664. [Google Scholar] [CrossRef]
- Tomic, M.T.; Farr-Jones, S.; Syar, E.S.; Niemuth, N.; Kobs, D.; Hackett, M.J.; Espinoza, Y.; Martinez, Z.; Pham, K.; Snow, D.M.; et al. Neutralizing Concentrations of Anti-Botulinum Toxin Antibodies Positively Correlate with Mouse Neutralization Assay Results in a Guinea Pig Model. Toxins 2021, 13, 671. [Google Scholar] [CrossRef] [PubMed]
- Liljeqvist, S.; Stahl, S. Production of recombinant subunit vaccines: Protein immunogens, live delivery systems and nucleic acid vaccines. J. Biotechnol. 1999, 73, 1–33. [Google Scholar] [CrossRef]
- Mabrouk, M.T.; Huang, W.C.; Deng, B.; Li-Purcell, N.; Seffouh, A.; Ortega, J.; Ekin Atilla-Gokcumen, G.; Long, C.A.; Miura, K.; Lovell, J.F. Lyophilized, antigen-bound liposomes with reduced MPLA and enhanced thermostability. Int. J. Pharm. 2020, 589, 119843. [Google Scholar] [CrossRef]
- Goldstein, N.; Bockstal, V.; Bart, S.; Luhn, K.; Robinson, C.; Gaddah, A.; Callendret, B.; Douoguih, M. Safety and Immunogenicity of Heterologous and Homologous 2-Dose Regimens of Adenovirus Serotype 26- and Modified Vaccinia Ankara-Vectored Ebola Vaccines: A Randomized, Controlled Phase 1 Study. J. Infect. Dis. 2022, 226, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Ura, T.; Okuda, K.; Shimada, M. Developments in Viral Vector-Based Vaccines. Vaccines 2014, 2, 624–641. [Google Scholar] [CrossRef]
- Casimiro, D.R.; Bett, A.J.; Fu, T.M.; Davies, M.E.; Tang, A.; Wilson, K.A.; Chen, M.; Long, R.; McKelvey, T.; Chastain, M.; et al. Heterologous human immunodeficiency virus type 1 priming-boosting immunization strategies involving replication-defective adenovirus and poxvirus vaccine vectors. J. Virol. 2004, 78, 11434–11438. [Google Scholar] [CrossRef]
- Xu, S.; Yang, K.; Li, R.; Zhang, L. mRNA Vaccine Era-Mechanisms, Drug Platform and Clinical Prospection. Int. J. Mol. Sci. 2020, 21, 6582. [Google Scholar] [CrossRef]
- Jackson, N.A.C.; Kester, K.E.; Casimiro, D.; Gurunathan, S.; DeRosa, F. The promise of mRNA vaccines: A biotech and industrial perspective. NPJ Vaccines 2020, 5, 11. [Google Scholar] [CrossRef]
- Hotez, P.J.; Bottazzi, M.E. Developing a low-cost and accessible COVID-19 vaccine for global health. PLoS Neglected Trop. Dis. 2020, 14, e0008548. [Google Scholar] [CrossRef]
- Crommelin, D.J.A.; Anchordoquy, T.J.; Volkin, D.B.; Jiskoot, W.; Mastrobattista, E. Addressing the Cold Reality of mRNA Vaccine Stability. J. Pharm. Sci. 2021, 110, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, H.; Lam, K.; Bajusz, C.; Laczko, D.; Kariko, K.; Schreiner, P.; Martin, A.; Lutwyche, P.; Heyes, J.; Pardi, N. Lyophilization provides long-term stability for a lipid nanoparticle-formulated, nucleoside-modified mRNA vaccine. Mol. Ther. 2022, 30, 1941–1951. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vaccine Type | Advantages | Disadvantages |
|---|---|---|
| Live vaccines (attenuated) |
|
|
| Killed vaccines |
|
|
| Toxoids |
|
|
| Subunit vaccines |
|
|
| Recombinant toxin vaccines |
|
|
| mRNA vaccines |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, S.; Pellett, S. Recent Developments in Vaccine Design: From Live Vaccines to Recombinant Toxin Vaccines. Toxins 2023, 15, 563. https://doi.org/10.3390/toxins15090563
Gupta S, Pellett S. Recent Developments in Vaccine Design: From Live Vaccines to Recombinant Toxin Vaccines. Toxins. 2023; 15(9):563. https://doi.org/10.3390/toxins15090563
Chicago/Turabian StyleGupta, Sonal, and Sabine Pellett. 2023. "Recent Developments in Vaccine Design: From Live Vaccines to Recombinant Toxin Vaccines" Toxins 15, no. 9: 563. https://doi.org/10.3390/toxins15090563
APA StyleGupta, S., & Pellett, S. (2023). Recent Developments in Vaccine Design: From Live Vaccines to Recombinant Toxin Vaccines. Toxins, 15(9), 563. https://doi.org/10.3390/toxins15090563