Mechanisms of Cisplatin Nephrotoxicity

Abstract
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
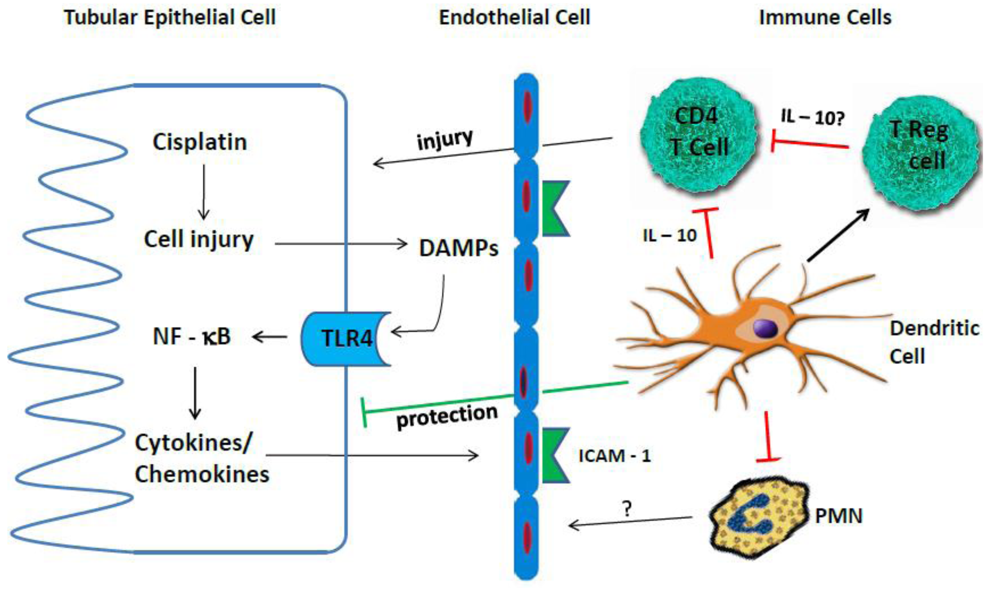{kind=link}
| Acute kidney injury (20–30%) | [15,16] |
| Hypomagnesemia (40–100%) | [17,18,19,20,21] |
| Fanconi-like syndrome | [22,23,24,25,26] |
| Distal renal tubular acidosis | [27] |
| Hypocalcemia | [28,29] |
| Renal salt wasting | [22,30,31,32,33,34,35,36] |
| Renal concentrating defect | [22,34,37,38,39,40] |
| Hyperuricemia | [41] |
| Transient proteinuria | [42] |
| Erythropoietin deficiency | [43] |
| Thrombotic microangiopathy | [44] |
| Chronic renal failure | [15,45,46] |
2. Clinical Characteristics of Cisplatin Nephrotoxicity
| Increased risk |
| Dose |
| Frequency |
| Cumulative dose |
| Older age |
| Female sex |
| Smoking |
| Hypoalbuminemia |
| Pre-existing renal insufficiency (limited data in humans) |
| Decreased risk |
| Diabetes (uncertain in humans) |
| OCT2 polymorphisms |
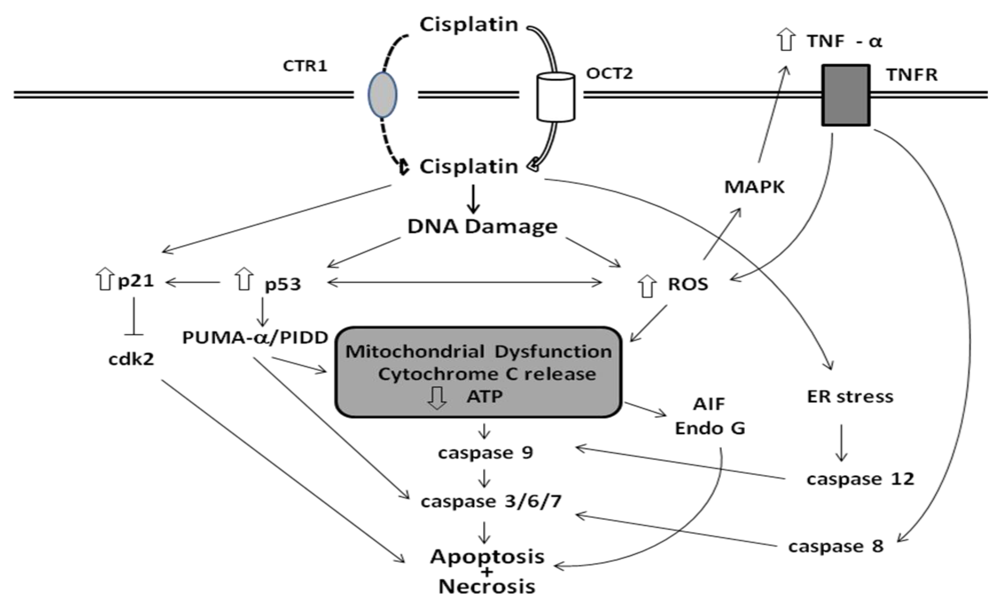
3. Mechanisms of Cisplatin Nephrotoxicity
3.1. Accumulation of Cisplatin in Kidney Cells
3.2. Biotransformation of Cisplatin in the Kidney
3.3. Cellular Targets of Cisplatin

3.4. Apoptotic Pathways of Cisplatin Cytotoxicity
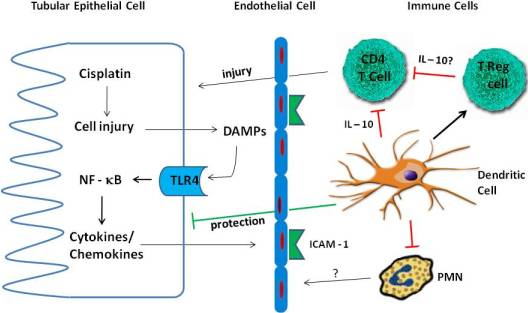
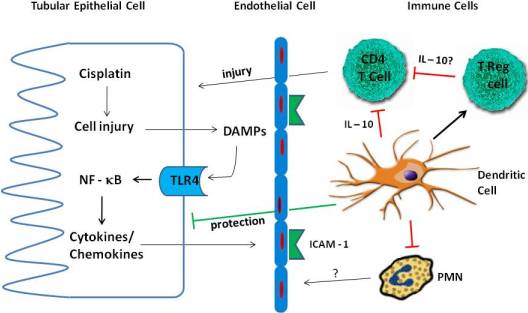
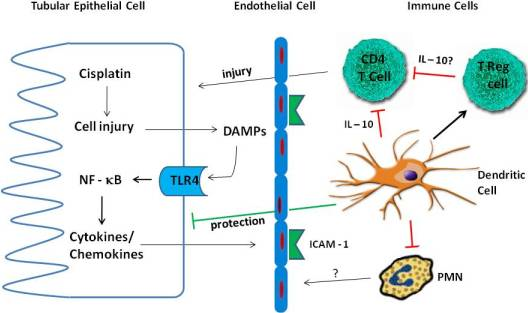
4. Inflammation in Cisplatin Nephrotoxicity

4.1. Cytokines
4.2. TLR Receptors
4.3. Immune Cells
4.3.1. Neutrophils
4.3.2. Macrophages
4.3.3. T Cells
4.3.4. Treg Cells
4.3.5. Dendritic Cells
5. Prevention of Cisplatin Nephrotoxicity
| Reduced renal cisplatin accumulation or activation | |
| OCT2 inhibitors, e.g., cimetidine or metformin | [61,208] |
| Ctr1 inhibitors, e.g., copper | [68] |
| Micellar/liposomal cisplatin | [206,207] |
| Gamma-glutamyl transpeptidase inhibitors | [76,210] |
| Glutathione transferase inhibitors | [74] |
| Anti-oxidants | |
| Amifostine | [203] |
| BNP7787 | [211] |
| N-acetyl cysteine | [212] |
| Superoxide dismutase | [23,146] |
| Catalase | [149] |
| Selenium and Vitamin E | [150] |
| Heme oxygenase-1 induction | [96] |
| Iron chelators, e.g., Desferoximine | [145] |
| Allopurinol plus ebselen | [213] |
| Milk thistle extract (silymarin) | [214] |
| Cannabidiol | [215] |
| Lycopene | [216] |
| Anti-apoptosis | |
| p53 inhibitors, e.g., pifithrin | [100,115,127,128,129] |
| HDAC inhibitors | [137,138] |
| Caspase inhibitors | [113] |
| p21agonists/CDK2 inhibitors | [123,124] |
| Anti-inflammation | |
| TNF-α antagonists | [102] |
| TLR4 antagonists | [162] |
| p38 inhibitors | [142] |
| JNK inhibitors | [141] |
| Salicylates | [98] |
| PPAR-α ligands, e.g., fibrates | [217] |
| PPAR-γ ligands, e.g. rosiglitazone | [218] |
| Alpha lipoic acid | [219] |
| IL-10 | [152] |
6. Summary
Acknowledgements
References
- Hartmann, J.T.; Fels, L.M.; Knop, S.; Stolt, H.; Kanz, L.; Bokemeyer, C. A randomized trial comparing the nephrotoxicity of cisplatin/ifosfamide-based combination chemotherapy with or without amifostine in patients with solid tumors. Invest. New Drugs 2000, 18, 281–289. [Google Scholar]
- Hartmann, J.T.; Lipp, H.-P. Toxicity of platinum compounds. Expert Opin. Pharmacother. 2003, 4, 889–901. [Google Scholar]
- Sastry, J.; Kellie, S.J. Severe neurotoxicity, ototoxicity and nephrotoxicity following high-dose cisplatin and amifostine. Pediatr. Hematol. Oncol. 2005, 22, 441–445. [Google Scholar]
- Arany, I.; Safirstein, R.L. Cisplatin nephrotoxicity. Semin. Nephrol. 2003, 23, 460–464. [Google Scholar]
- Boulikas, T. Poly(ADP-ribose) synthesis in blocked and damaged cells and its relation to carcinogens. Anticancer Res. 1992, 12, 885–898. [Google Scholar]
- Planting, A.; Catimel, G.; de Mulder, P.; de Graeff, A.; Hoppener, F.; Verweij, J.; Oster, W.; Vermorken, J. Randomized study of a short course of weekly cisplatin with or without amifostine in advanced head and neck cancer. Ann. Oncol. 1999, 10, 693–700. [Google Scholar] [Green Version]
- Loehrer, P.J.; Gonin, R.; Nichols, C.R.; Weathers, T.; Einhorn, L.H. Vinblastine plus ifosphamide plus cisplatin as initial salvage therapy in recurrent germ cell tumor. J. Clin. Oncol. 1998, 16, 2500–2504. [Google Scholar]
- Noda, K.; Nishiwaki, Y.; Kawahara, M.; Negoro, S.; Sugiura, T.; Yokoyama, A.; Saijo, N. Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small-cell lung cancer. N. Engl. J. Med. 2002, 346, 85–91. [Google Scholar]
- Gatzemeier, U.; von Pawel, J.; ten Velde, G.; Mattson, K.; DeMarinis, F.; Harper, P.; Salvati, F.; Robinet, G.; Lucenti, A.; Bogarerts, J.; Gallant, G. Phase III comparative study of high-dose cisplatin versus a combination of paclitaxel and cisplatin in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2000, 18, 3390–3399. [Google Scholar]
- Bolis, G.; Favalli, G.; Danese, S.; Zanaboni, F.; Mangili, G.; Scarabelli, C.; Tateo, S.; Valsecchi, M.; Scarfone, G.; Richiardi, G.; Frigerio, L.; Melpignano, M.; Villa, A.; Parazzini, F. Weekly cisplatin given for 2 months versus cisplatin plus cyclophosphamide given for 5 months after cytoreductive surgery for advanced ovarian cancer. J. Clin. Oncol. 1997, 15, 1938–1944. [Google Scholar]
- Hoskins, P.; Eisenhauer, E.; Vergote, I.; Dubuc-Lissoir, J.; Fisher, B.; Grimshaw, R.; Oza, A.; Plante, M.; Stuart, G.; Vermorken, J. Phase II feasibility study of sequential couplets of Cisplatin/Topotecan followed by paciltaxel/cisplatin as primary treatment of advanced epithelial ovarian cancer: A National Cancer Institute of Canada Clinical Trials Group study. J. Clin. Oncol. 2000, 18, 4038–4044. [Google Scholar]
- Rose, P.; Bundy, B.; Watkins, E.; Thigpen, J.; Deppe, G.; Maiman, M.; Clarke-Pearson, D.; Insalaco, S. Concurrent cisplatin-based radiotherapy and chemotherapy for locally advanced cervical cancer. N. Engl. J. Med. 1999, 340, 1144–1153. [Google Scholar]
- Coppin, C.; Gospodarowicz, M.; James, K.; Tannock, I.; Zee, B.; Carson, J.; Peter, J.; Sullivan, D. Improved local control of invasive bladder cancer by concurrent cisplatin and preoperative or definitive radiation. J. Clin. Oncol. 1996, 14, 2901–2907. [Google Scholar]
- Pritchard, J.; Brown, J.; Shafford, E.; Perilongo, G.; Brock, P.; Dicks-Mireaux, C.; Keeling, J.; Phillips, A.; Vos, A.; Plaschkes, J. Cisplatin, doxorubicin, and delayed surgery for childhood hepatoblastoma: A successful approach—results of the first prospective study of the Internal Society of Pediatric Oncology. J. Clin. Oncol. 2000, 18, 3819–3828. [Google Scholar]
- Madias, N.E.; Harrington, J.T. Platinum nephrotoxicity. Am. J. Med. 1978, 65, 307–314. [Google Scholar]
- Goldstein, R.S.; Mayor, G.H. Minireview. The nephrotoxicity of cisplatin. Life Sci. 1983, 32, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Schilsky, R.L.; Anderson, T.O.M. Hypomagnesemia and renal magnesium wasting in patients receiving cisplatin. Ann. Intern. Med. 1979, 90, 929–931. [Google Scholar]
- Lam, M.; Adelstein, D.J. Hypomagnesemia and renal magnesium wasting in patients treated with cisplatin. Am. J. Kidney Dis. 1986, 8, 164–169. [Google Scholar]
- Ries, F.; Klastersky, J. Nephrotoxicity induced by cancer chemotherapy with special emphasis on cisplatin toxicity. Am. J. Kidney Dis. 1986, 13, 368–379. [Google Scholar]
- Sutton, R.A.; Walker, V.R.; Halabe, A.; Swenerton, K.; Coppin, C.M. Chronic hypomagnesemia caused by cisplatin: Effect of calcitriol. J. Lab. Clin. Med. 1991, 117, 40–43. [Google Scholar]
- Parfitt, A.; Kleerekoper, M. Clinical disorders of calcium, phosphorous and magnesium metabolism. In Clinical Disorders of Fluid and Electrolyte Metabolism; Maxwell, M., Kleeman, C., Eds.; McGraw-Hill: New York, NY, USA,, 1980. [Google Scholar]
- Kim, Y.K.; Byun, H.S.; Kim, Y.H.; Woo, J.S.; Lee, S.H. Effect of Cisplatin on renal-function in rabbits: Mechanism of reduced glucose reabsorption. Toxicol. Appl. Pharmacol. 1995, 130, 19–26. [Google Scholar]
- Wangila, G.W.; Nagothu, K.K.; Steward, R., III; Bhatt, R.; Iyere, P.A.; Willingham, W.M.; Sorenson, J.R.; Shah, S.V.; Portilla, D. Prevention of cisplatin-induced kidney epithelial cell apoptosis with a Cu superoxide dismutase-mimetic [copper2II(3,5-ditertiarybutylsalicylate)4(ethanol)4]. Toxicol. In Vitro 2006, 20, 1300–1312. [Google Scholar]
- Goldstein, R.S.; Mayor, G.H.; Rosenbaum, R.W.; Hook, J.B.; Santiago, J.V.; Bond, J.T. Glucose intolerance following cis-platinum treatment in rats. Toxicology 1982, 24, 273–280. [Google Scholar]
- Goldstein, R.S.; Mayor, G.H.; Gingerich, R.L.; Hook, J.B.; Rosenbaum, R.W.; Bond, J.T. The effects of cisplatin and other divalent platinum compounds on glucose metabolism and pancreatic endocrine function. Toxicol. Appl. Pharmacol. 1983, 69, 432–441. [Google Scholar]
- Oeffinger, K.; Hudson, M. Long-term complications following childhood and adolescent cancer: Foundations for providing risk-based health care for survivors. CA Cancer J. Clin. 2004, 54, 208–236. [Google Scholar]
- Swainson, C.P.; Colls, B.M.; Fitzharris, B.M. Cis-platinum and distal renal tubule toxicity. N. Z. Med. J. 1985, 98, 375–378. [Google Scholar]
- Suh, S.M.; Tashjian, A.H.; Matsuo, N.; Parkinson, D.K.; Fraser, D. Pathogenesis of hypocalcemia in primary hypomagnesemia: Normal end-organ responsiveness to parathyroid hormone, impaired parathyroid gland function. J. Clin. Invest. 1973, 52, 153–160. [Google Scholar]
- Connor, T.B.; Toskes, P.; Mahaffey, J.; Martin, L.G.; Williams, J.B.; Walser, M. Parathyroid function during chronic magnesium deficiency. Johns Hopkins Med. J. 1972, 131, 100–117. [Google Scholar]
- Hutchison, F.N.; Perez, E.A.; Gandara, D.R.; Lawrence, H.J.; Kaysen, G.A. Renal salt wasting in patients treated with cisplatin. Ann. Intern. Med. 1988, 108, 21–25. [Google Scholar]
- Cao, L.M.D.; Joshi, P.M.D.; Sumoza, D.M.D. Renal salt-wasting syndrome in a patient with cisplatin-induced hyponatremia: Case report. Am. J. Clin. Oncol. 2002, 25, 344–346. [Google Scholar]
- Kurtzberg, J.; Dennis, V.W.; Kinney, T.R. Cisplatinum-induced renal salt wasting. Med. Pediatr. Oncol. 1984, 12, 150–154. [Google Scholar]
- Shakhov, A.; Collart, M.; Vassalli, P.; Nedospasov, S.; Jongeneel, C. kB-Type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor α gene in primary macrophages. J. Exp. Med. 1990, 171, 35–47. [Google Scholar]
- Seguro, A.C.; Shimizu, M.H.; Kudo, L.H.; dos Santos Rocha, A. Renal concentration defect induced by cisplatin. The role of thick ascending limb and papillary collecting duct. Am. J. Nephrol. 1989, 9, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Lammers, P.J.; White, L.; Ettinger, L.J. Cis-platinum-induced renal sodium wasting. Med. Pediatr. Oncol. 1984, 12, 343–346. [Google Scholar]
- Tscherning, C.; Hervé, R.; Chancholle, A.; Claeyssens, S.; Robert, A.; Fabre, J.; Bouissou, F. Recurrent renal salt wasting in a child treated with carboplatin and etoposide. Cancer 1994, 73, 1761–1763. [Google Scholar]
- Safirstein, R.; Miller, P.; Dikman, S.; Lyman, N.; Shapiro, C. Cisplatin nephrotoxicity in rats: Defect in papillary hypertonicity. Am. J. Physiol. Renal Physiol. 1981, 241, F175–F185. [Google Scholar]
- Gordon, J.A.; Peterson, L.N.; Anderson, R.J. Water metabolism after cisplatin in the rat. Am. J. Physiol. Renal Physiol. 1982, 243, F36–F43. [Google Scholar]
- Safirstein, R.; Zelent, A.; Gordon, R. Cisplatin nephrotoxicity: New insights into mechanism. In Organ Directed Toxicities of Anticancer Drugs, First International Symposium on the Organ Directed Toxicities of Anticancer Drugs, Burlington, VT, USA, June 4–6, 1987; Hacker, M.P., Lazo, J.S., Tritton, T.R., Eds.; Martinus Nijhoff Publishers: Boston, MA, USA, 1988; p. 174. [Google Scholar]
- Launay-Vacher, V.; Rey, J.B.; Isnard-Bagnis, C.; Deray, G.; Daouphars, M. Prevention of cisplatin nephrotoxicity: State of the art and recommendations from the European Society of Clinical Pharmacy Special Interest Group on Cancer Care. Cancer Chemother. Pharmacol. 2008, 61, 903–909. [Google Scholar]
- Nanji, A.A.; Mikhael, N.Z.; Stewart, D.J. Increase in serum uric acid level associated with cisplatin therapy: Correlation with liver but not kidney platinum concentrations. Arch. Intern. Med. 1985, 145, 2013–2014. [Google Scholar]
- DeConti, R.C.; Toftness, B.R.; Lange, R.C.; Creasey, W.A. Clinical and pharmacological studies with cis-Diamminedichloroplatinum(II). Cancer Res. 1973, 33, 1310–1315. [Google Scholar]
- Wood, P.A.; Hrushesky, W.J. Cisplatin-associated anemia: An erythropoietin deficiency syndrome. J. Clin. Invest. 1995, 95, 1650–1659. [Google Scholar]
- Jackson, A.M.; Rose, B.D.; Graff, L.G.; Jacobs, J.B.; Schwartz, J.H.; Strauss, G.M.; Yang, J.P.S.; Rudnick, M.R.; Elfenbein, I.B.; Narins, R.G. Thrombotic microangiopathy and renal failure associated with antineoplastic chemotherapy. Ann. Intern. Med. 1984, 101, 41–44. [Google Scholar]
- Brillet, G.; Deray, G.; Jacquiaud, C.; Mignot, L.; Bunker, D.; Meillet, D.; Raymond, F.; Jacobs, C. Long-term renal effect of cisplatin in man. Am. J. Nephrol. 1994, 14, 81–84. [Google Scholar]
- Koch Nogueira, P.C.; Hadj-Aïssa, A.; Schell, M.; Dubourg, L.; Brunat-Mentigny, M.; Cochat, P. Long-term nephrotoxicity of cisplatin, ifosfamide, and methotrexate in osteosarcoma. Pediatr. Nephrol. 1998, 12, 572–575. [Google Scholar]
- Rosenberg, B.; Vancamp, L.; Krigas, T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature 1965, 205, 698–699. [Google Scholar]
- Rosenberg, B.; VanCamp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar]
- Kociba, R.J.; Sleight, S.D. Acute toxicologic and pathologic effects of cis-diamminedichloroplatinum (NSC-119875) in the male rat. Cancer Chemother. Rep. 1971, 55, 1–8. [Google Scholar]
- Hartmann, J.T.; Kollmannsberger, C.; Kanz, L.; Bokemeyer, C. Platinum organ toxicity and possible prevention in patients with testicular cancer. Int. J. Cancer 1999, 83, 866–869. [Google Scholar]
- Santoso, J.T.; Lucci, J.A., III; Coleman, R.L.; Schafer, I.; Hannigan, E.V. Saline, mannitol, and furosemide hydration in acute cisplatin nephrotoxicity: A randomized trial. Cancer Chemother. Pharmacol. 2003, 52, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Nazneen, A.; Abid, M.R.; Razzaque, M.S. Cisplatin-associated nephrotoxicity and pathological events. Contrib. Nephrol. 2005, 148, 107–121. [Google Scholar]
- Reece, P.A.; Stafford, I.; Russell, J.; Khan, M.; Gill, P.G. Creatinine clearance as a predictor of ultrafilterable platinum disposition in cancer patients treated with cisplatin: Relationship between peak ultrafilterable platinum plasma levels and nephrotoxicity. J. Clin. Oncol. 1987, 5, 304–309. [Google Scholar]
- de Jongh, F.E.; Verweij, J.; Loos, W.J.; de Wit, R.; de Jonge, M.J.A.; Planting, A.S.T.; Nooter, K.; Stoter, G.; Sparreboom, A. Body-surface area-based dosing does not increase accuracy of predicting cisplatin exposure. J. Clin. Oncol. 2001, 19, 3733–3739. [Google Scholar]
- de Jongh, F.E.; van Veen, R.N.; Veltman, S.J.; de Wit, R.; van der Burg, M.E.L.; van den Bent, M.J.; Planting, A.; Graveland, W.J.; Stoter, G.; Verweij, J. Weekly high-dose cisplatin is a feasible treatment option: Analysis on prognostic factors for toxicity in 400 patients. Br. J. Cancer 2003, 88, 1199–1206. [Google Scholar]
- Raj, G.V.; Iasonos, A.; Herr, H.; Donat, S.M. Formulas calculating creatinine clearance are inadequate for determining eligibility for cisplatin-based chemotherapy in bladder cancer. J. Clin. Oncol. 2006, 24, 3095–3100. [Google Scholar]
- Scott, L.A.; Madan, E.; Valentovic, M.A. Attenuation of cisplatin nephrotoxicity by streptozotocin-induced diabetes. Fundam. Appl. Toxicol. 1989, 12, 530–539. [Google Scholar]
- Gogas, H.; Shapiro, F.; Aghajanian, C.; Fennelly, D.; Almadrones, L.; Hoskins, W.J.; Spriggs, D.R. The impact of diabetes mellitus on the toxicity of therapy for advanced ovarian cancer. Gynecol. Oncol. 1996, 61, 22–26. [Google Scholar]
- Stewart, D.J.; Dulberg, C.S.; Mikhael, N.Z.; Redmond, M.D.; Montpetit, V.A.; Goel, R. Association of cisplatin nephrotoxicity with patient characteristics and cisplatin administration methods. Cancer Chemother. Pharmacol. 1997, 40, 293–308. [Google Scholar]
- Filipski, K.K.; Mathijssen, R.H.; Mikkelsen, T.S.; Schinkel, A.H.; Sparreboom, A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin. Pharmacol. Ther. 2009, 86, 396–402. [Google Scholar]
- Ciarimboli, G.; Deuster, D.; Knief, A.; Sperling, M.; Holtkamp, M.; Edemir, B.; Pavenstadt, H.; Lanvers-Kaminsky, C.; am Zehnhoff-Dinnesen, A.; Schinkel, A.H.; Koepsell, H.; Jurgens, H.; Schlatter, E. Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions. Am. J. Pathol. 2010, 176, 1169–1180. [Google Scholar]
- Yao, X.; Panichpisal, K.; Kurtzman, N.; Nugent, K. Cisplatin nephrotoxicity: A review. Am. J. Med. Sci. 2007, 334, 115–124. [Google Scholar]
- Safirstein, R.; Miller, P.; Guttenplan, J.B. Uptake and metabolism of cisplatin by rat kidney. Kidney Int. 1984, 25, 753–758. [Google Scholar]
- Endo, T.; Kimura, O.; Sakata, M. Carrier-mediated uptake of cisplatin by the OK renal epithelial cell line. Toxicology 2000, 146, 187–195. [Google Scholar]
- Kolb, R.; Ghazi, M.; Barfuss, D. Inhibition of basolateral transport and cellular accumulation of cDDP and N-acetyl-L-cysteine-cDDP by TEA and PAH in the renal proximal tubule. Cancer Chemother. Pharmacol. 2003, 51, 132–138. [Google Scholar]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar]
- Holzer, A.K.; Samimi, G.; Katano, K.; Naerdemann, W.; Lin, X.; Safaei, R.; Howell, S.B. The copper influx transporter human copper transport protein 1 regulates the uptake of cisplatin in human ovarian carcinoma cells. Mol. Pharmacol. 2004, 66, 817–823. [Google Scholar]
- Pabla, N.; Murphy, R.F.; Liu, K.; Dong, Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am. J. Physiol. Renal Physiol. 2009, 296, F505–F511. [Google Scholar]
- Ludwig, T.; Riethmuller, C.; Gekle, M.; Schwerdt, G.; Oberleithner, H. Nephrotoxicity of platinum complexes is related to basolateral organic cation transport. Kidney Int. 2004, 66, 196–202. [Google Scholar]
- Ciarimboli, G.; Ludwig, T.; Lang, D.; Pavenstadt, H.; Koepsell, H.; Piechota, H.J.; Haier, J.; Jaehde, U.; Zisowsky, J.; Schlatter, E. Cisplatin nephrotoxicity is critically mediated via the human organic cation transporter 2. Am. J. Pathol. 2005, 167, 1477–1484. [Google Scholar]
- Filipski, K.K.; Loos, W.J.; Verweij, J.; Sparreboom, A. Interaction of Cisplatin with the human organic cation transporter 2. Clin. Cancer Res. 2008, 14, 3875–3880. [Google Scholar]
- Yonezawa, A.; Masuda, S.; Yokoo, S.; Katsura, T.; Inui, K.-i. Cisplatin and oxaliplatin, but not carboplatin and nedaplatin, are substrates for human organic cation transporters (SLC22A1-3 and multidrug and toxin extrusion family). J. Pharmacol. Exp. Ther. 2006, 319, 879–886. [Google Scholar]
- Townsend, D.M.; Tew, K.D.; He, L.; King, J.B.; Hanigan, M.H. Role of glutathione S-transferase Pi in cisplatin-induced nephrotoxicity. Biomed. Pharmacother. 2009, 63, 79–85. [Google Scholar]
- Sadzuka, Y.; Shimizu, Y.; Takino, Y.; Hirota, S. Protection against cisplatin-induced nephrotoxicity in the rat by inducers and an inhibitor of glutathione S-transferase. Biochem. Pharmacol. 1994, 48, 453–459. [Google Scholar]
- Townsend, D.M.; Deng, M.; Zhang, L.; Lapus, M.G.; Hanigan, M.H. Metabolism of Cisplatin to a nephrotoxin in proximal tubule cells. J. Am. Soc. Nephrol. 2003, 14, 1–10. [Google Scholar]
- Townsend, D.M.; Hanigan, M.H. Inhibition of gamma-glutamyl transpeptidase or cysteine S-conjugate beta-lyase activity blocks the nephrotoxicity of cisplatin in mice. J. Pharmacol. Exp. Ther. 2002, 300, 142–148. [Google Scholar]
- Zhang, L.; Hanigan, M.H. Role of cysteine S-conjugate β-lyase in the metabolism of cisplatin. J. Pharmacol. Exp. Ther. 2003, 306, 988–994. [Google Scholar]
- Pascoe, J.M.; Roberts, J.J. Interactions between mammalian cell DNA and inorganic platinum compounds. I. DNA interstrand cross-linking and cytotoxic properties of platinum(II) compounds. Biochem. Pharmacol. 1974, 23, 1359–1365. [Google Scholar] [PubMed]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar]
- Burger, H.; Nooter, K.; Boersma, A.W.M.; Kortland, C.J.; Stoter, G. Lack of correlation between cisplatin-induced apoptosis, p53 status and expression of Bcl-2 family proteins in testicular germ cell tumour cell lines. Int. J. Cancer 1997, 73, 592–599. [Google Scholar]
- Mandic, A.; Hansson, J.; Linder, S.; Shoshan, M.C. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J. Biol. Chem. 2003, 278, 9100–9106. [Google Scholar]
- Cullen, K.J.; Yang, Z.; Schumaker, L.; Guo, Z. Mitochondria as a critical target of the chemotheraputic agent cisplatin in head and neck cancer. J. Bioenerg. Biomembr. 2007, 39, 43–50. [Google Scholar]
- Qian, W.; Nishikawa, M.; Haque, A.M.; Hirose, M.; Mashimo, M.; Sato, E.; Inoue, M. Mitochondrial density determines the cellular sensitivity to cisplatin-induced cell death. Am. J. Physiol. Cell Physiol. 2005, 289, C1466–C1475. [Google Scholar]
- Hirama, M.; Isonishi, S.; Yasuda, M.; Ishikawa, H. Characterization of mitochondria in cisplatin-resistant human ovarian carcinoma cells. Oncol. Rep. 2006, 16, 997–1002. [Google Scholar]
- Gullans, S.R.; Mandel, L.J. Coupling of energy transport in proximal and distal nephron. In The Kidney: Physiology and Pathophysiology, 3rd; Seldin, D.W., Giebisch, G., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2000; Volume 1, pp. 445–482. [Google Scholar]
- Olivero, O.A.; Chang, P.K.; LopezLarraza, D.M.; SeminoMora, M.C.; Poirier, M.C. Preferential formation and decreased removal of cisplatin-DNA adducts in Chinese hamster ovary cell mitochondrial DNA as compared to nuclear DNA. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 1997, 391, 79–86. [Google Scholar]
- Li, S.; Wu, P.; Yarlagadda, P.; Vadjunec, N.M.; Proia, A.D.; Harris, R.A.; Portilla, D. PPAR alpha ligand protects during cisplatin-induced acute renal failure by preventing inhibition of renal FAO and PDC activity. Am. J. Physiol. Renal Physiol. 2004, 286, F572–F580. [Google Scholar]
- Portilla, D.; Dai, G.; McClure, T.; Bates, L.; Kurten, R.; Megyesi, J.; Price, P.; Li, S. Alterations of PPARalpha and its coactivator PGC-1 in cisplatin-induced acute renal failure. Kidney Int. 2002, 62, 1208–1218. [Google Scholar]
- Li, S.; Basnakian, A.; Bhatt, R.; Megyesi, J.; Gokden, N.; Shah, S.V.; Portilla, D. PPAR-alpha ligand ameliorates acute renal failure by reducing cisplatin-induced increased expression of renal endonuclease G. Am. J. Physiol. Renal Physiol. 2004, 287, F990–F998. [Google Scholar]
- Kruidering, M.; Van de Water, B.; de Heer, E.; Mulder, G.J.; Nagelkerke, J.F. Cisplatin-induced nephrotoxicity in porcine proximal tubular cells: Mitochondrial dysfunction by inhibition of complexes I to IV of the respiratory chain. J. Pharmacol. Exp. Ther. 1997, 280, 638–649. [Google Scholar]
- Arany, I.; Kaushal, G.P.; Portilla, D.; Megyesi, J.; Price, P.M.; Safirstein, R.L. Cellular mechanisms of nephrotoxicity. In Clinical Nephrotoxins; Broe, M.E.D., Porter, G.A., Bennett, W.M., Deray, G., Eds.; Springer: New York, NY, USA, 2008; pp. 155–170. [Google Scholar]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar]
- Jiang, M.; Dong, Z. Regulation and pathological role of p53 in cisplatin nephrotoxicity. J. Pharmacol. Exp. Ther. 2008, 327, 300–307. [Google Scholar]
- Lieberthal, W.; Triaca, V.; Levine, J. Mechanisms of death induced by cisplatin in proximal tubular eipthelial cells: Apoptosis vs. necrosis. Am. J. Physiol. Renal Physiol. 1996, 270, F700–F708. [Google Scholar]
- Lee, R.H.; Song, J.M.; Park, M.Y.; Kang, S.K.; Kim, Y.K.; Jung, J.S. Cisplatin-induced apoptosis by translocation of endogenous Bax in mouse collecting duct cells. Biochem. Pharmacol. 2001, 62, 1013–1023. [Google Scholar]
- Shiraishi, F.; Curtis, L.; Truong, L.; Poss, K.; Visner, G.; Madsen, K.; Nick, H.; Agarwal, A. Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am. J. Physiol. Renal Physiol. 2000, 278, F726–F736. [Google Scholar]
- Megyesi, J.; Safirstein, R.L.; Price, P.M. Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the course of cisplatin-induced acute renal failure. J. Clin. Invest. 1998, 101, 777–782. [Google Scholar]
- Ramesh, G.; Reeves, W.B. Salicylate reduces cisplatin nephrotoxicity by inhibition of tumor necrosis factor-α. Kidney Int. 2004, 65, 490–498. [Google Scholar]
- Tsuruya, K.; Ninomiya, T.; Tokumoto, M.; Hirakawa, M.; Matsutani, K.; Taniguchi, M.; Fukuda, K.; Kanai, H.; Kishihara, K.; Hirakata, H.; Iida, M. Direct involvement of the receptor-mediated apoptotic pathways in cisplatin-induced renal tubular cell death. Kidney Int. 2003, 63, 72–82. [Google Scholar]
- Seth, R.; Yang, C.; Kaushal, V.; Shah, S.V.; Kaushal, G.P. p53-dependent caspase-2 activation in mitochondrial release of apoptosis-inducing factor and its role in renal tubular epithelial cell injury. J. Biol. Chem. 2005, 280, 31230–31239. [Google Scholar]
- Takeda, M.; Kobayashi, M.; Shirato, I.; Osaki, T.; Endou, H. Cisplatin-induced apoptosis of immortalized mouse proximal tubule cells is mediated by interleukin-1 beta converting enzyme (ICE) family of proteases but inhibited by overexpression of Bcl-2. Arch. Toxicol. 1997, 71, 612–621. [Google Scholar]
- Ramesh, G.; Reeves, W.B. TNF-α mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J. Clin. Invest. 2002, 110, 835–842. [Google Scholar]
- Ramesh, G.; Reeves, W.B. TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. Am. J. Physiol. Renal Physiol. 2003, 285, F610–F618. [Google Scholar]
- Cilenti, L.; Kyriazis, G.A.; Soundarapandian, M.M.; Stratico, V.; Yerkes, A.; Park, K.M.; Sheridan, A.M.; Alnemri, E.S.; Bonventre, J.V.; Zervos, A.S. Omi/HtrA2 protease mediates cisplatin-induced cell death in renal cells. Am. J. Physiol. Renal Physiol. 2005, 288, F371–F379. [Google Scholar]
- Park, M.S.; De Leon, M.; Devarajan, P. Cisplatin induces apoptosis in LLC-PK1 cells via activation of mitochondrial pathways. J. Am. Soc. Nephrol. 2002, 13, 858–865. [Google Scholar]
- Nagothu, K.K.; Bhatt, R.; Kaushal, G.P.; Portilla, D. Fibrate prevents cisplatin-induced proximal tubule cell death. Kidney Int. 2005, 68, 2680–2693. [Google Scholar]
- Jiang, M.; Wei, Q.; Wang, J.; Du, Q.; Yu, J.; Zhang, L.; Dong, Z. Regulation of PUMA-alpha by p53 in cisplatin-induced renal cell apoptosis. Oncogene 2006, 25, 4056–4066. [Google Scholar]
- Wei, Q.; Dong, G.; Franklin, J.; Dong, Z. The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int. 2007, 72, 53–62. [Google Scholar]
- Jiang, M.; Wang, C.-Y.; Huang, S.; Yang, T.; Dong, Z. Cisplatin-induced apoptosis in p53-deficient renal cells via the intrinsic mitochondrial pathway. Am. J. Physiol. Renal Physiol. 2009, 296, F983–F993. [Google Scholar]
- Yin, X.; Apostolov, E.O.; Shah, S.V.; Wang, X.; Bogdanov, K.V.; Buzder, T.; Stewart, A.G.; Basnakian, A.G. Induction of renal endonuclease G by cisplatin is reduced in DNase I-deficient mice. J. Am. Soc. Nephrol. 2007, 18, 2544–2553. [Google Scholar]
- Dursun, B.; He, Z.; Somerset, H.; Oh, D.J.; Faubel, S.; Edelstein, C.L. Caspases and calpain are independent mediators of cisplatin-induced endothelial cell necrosis. Am. J. Physiol. Renal Physiol. 2006, 291, F578–F587. [Google Scholar]
- Fukuoka, K.; Takeda, M.; Kobayashi, M.; Osaki, T.; Shirato, I.; Soejima, A.; Nagasawa, T.; Endou, H. Distinct interleukin-1beta-converting enzyme family proteases mediate cisplatin- and staurosporine-induced apoptosis of mouse proximal tubule cells. Life Sci. 1998, 62, 1125–1138. [Google Scholar]
- Kaushal, G.; Kaushal, V.; Hong, X.; Shah, S. Role and regulation of activation of caspases in cisplatin-induced injury to renal tubular epithelial cells. Kidney Int. 2001, 60, 1726–1736. [Google Scholar]
- Yang, C.; Kaushal, V.; Haun, R.S.; Seth, R.; Shah, S.V.; Kaushal, G.P. Transcriptional activation of caspase-6 and -7 genes by cisplatin-induced p53 and its functional significance in cisplatin nephrotoxicity. Cell Death Differ. 2008, 15, 530–544. [Google Scholar]
- Jiang, M.; Yi, X.; Hsu, S.; Wang, C.Y.; Dong, Z. Role of p53 in cisplatin-induced tubular cell apoptosis: Dependence on p53 transcriptional activity. Am. J. Physiol. Renal Physiol. 2004, 287, F1140–F1147. [Google Scholar]
- Liu, H.; Baliga, R. Endoplasmic reticulum stress-associated caspase 12 mediates cisplatin-induced LLC-PK1 cell apoptosis. J. Am. Soc. Nephrol. 2005, 16, 1985–1992. [Google Scholar]
- Cummings, B.S.; McHowat, J.; Schnellmann, R.G. Role of an endoplasmic reticulum Ca2+-independent phospholipase A2 in cisplatin-induced renal cell apoptosis. J. Pharmacol. Exp. Ther. 2004, 308, 921–928. [Google Scholar]
- Peyrou, M.; Hanna, P.E.; Cribb, A.E. Cisplatin, gentamicin, and p-aminophenol induce markers of endoplasmic reticulum stress in the rat kidneys. Toxicol. Sci. 2007, 99, 346–353. [Google Scholar]
- Kaushal, G.P.; Kaushal, V.; Herzog, C.; Yang, C. Autophagy delays apoptosis in renal tubular epithelial cells in cisplatin cytotoxicity. Autophagy 2008, 4, 710–712. [Google Scholar]
- Yang, C.; Kaushal, V.; Shah, S.V.; Kaushal, G.P. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am. J. Physiol. Renal Physiol. 2008, 294, F777–F787. [Google Scholar]
- Periyasamy-Thandavan, S.; Jiang, M.; Wei, Q.; Smith, R.; Yin, X.M.; Dong, Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 2008, 74, 631–640. [Google Scholar]
- Megyesi, J.; Udvarhelyi, N.; Safirstein, R.L.; Price, P.M. The p53-independent activation of transcription of p21WAF1/CIP1/SDI1 after acute renal failure. Am. J. Physiol. Renal Physiol. 1996, 271, F1211–F1216. [Google Scholar]
- Price, P.M.; Safirstein, R.L.; Megyesi, J. Protection of renal cells from cisplatin toxicity by cell cycle inhibitors. Am. J. Physiol. Renal Physiol. 2004, 286, F378–F384. [Google Scholar]
- Price, P.M.; Yu, F.; Kaldis, P.; Aleem, E.; Nowak, G.; Safirstein, R.L.; Megyesi, J. Dependence of cisplatin-induced cell death in vitro and in vivo on cyclin-dependent kinase 2. J. Am. Soc. Nephrol. 2006, 17, 2434–2442. [Google Scholar]
- Yu, F.; Megyesi, J.; Safirstein, R.L.; Price, P.M. Identification of the functional domain of p21(WAF1/CIP1) that protects cells from cisplatin cytotoxicity. Am. J. Physiol. Renal Physiol. 2005, 289, F514–F520. [Google Scholar]
- Bassett, E.A.; Wang, W.; Rastinejad, F.; El-Deiry, W.S. Structural and functional basis for therapeutic modulation of p53 signaling. Clin. Cancer Res. 2008, 14, 6376–6386. [Google Scholar]
- Wei, Q.; Dong, G.; Yang, T.; Megyesi, J.; Price, P.M.; Dong, Z. Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am. J. Physiol. Renal Physiol. 2007, 293, F1282–F1291. [Google Scholar]
- Cummings, B.S.; Schnellmann, R.G. Cisplatin-induced renal cell apoptosis: Caspase 3-dependent and -independent pathways. J. Pharmacol. Exp. Ther. 2002, 302, 8–17. [Google Scholar]
- Molitoris, B.A.; Dagher, P.C.; Sandoval, R.M.; Campos, S.B.; Ashush, H.; Fridman, E.; Brafman, A.; Faerman, A.; Atkinson, S.J.; Thompson, J.D.; Kalinski, H.; Skaliter, R.; Erlich, S.; Feinstein, E. siRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J. Am. Soc. Nephrol. 2009, 20, 1754–1764. [Google Scholar]
- Lebedeva, M.A.; Eaton, J.S.; Shadel, G.S. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 328–334. [Google Scholar]
- Yoshida, Y.; Izumi, H.; Torigoe, T.; Ishiguchi, H.; Itoh, H.; Kang, D.; Kohno, K. p53 physically interacts with mitochondrial transcription factor a and differentially regulates binding to damaged DNA. Cancer Res. 2003, 63, 3729–3734. [Google Scholar]
- Jiang, M.; Wei, Q.; Pabla, N.; Dong, G.; Wang, C.Y.; Yang, T.; Smith, S.B.; Dong, Z. Effects of hydroxyl radical scavenging on cisplatin-induced p53 activation, tubular cell apoptosis and nephrotoxicity. Biochem. Pharmacol. 2007, 73, 1499–1510. [Google Scholar]
- Pabla, N.; Huang, S.; Mi, Q.-S.; Daniel, R.; Dong, Z. ATR-Chk2 Signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis. J. Biol. Chem. 2008, 283, 6572–6583. [Google Scholar]
- Basnakian, A.G.; Apostolov, E.O.; Yin, X.; Napirei, M.; Mannherz, H.G.; Shah, S.V. Cisplatin nephrotoxicity is mediated by deoxyribonuclease I. J. Am. Soc. Nephrol. 2005, 16, 697–702. [Google Scholar]
- Yin, X.; Apostolov, E.O.; Shah, S.V.; Wang, X.; Bogdanov, K.V.; Buzder, T.; Stewart, A.G.; Basnakian, A.G. Induction of renal endonuclease G by cisplatin is reduced in DNase I-deficient mice. J. Am. Soc. Nephrol. 2007, 18, 2544–2553. [Google Scholar]
- Dong, G.; Wang, L.; Wang, C.-Y.; Yang, T.; Kumar, M.V.; Dong, Z. Induction of apoptosis in renal tubular cells by histone deacetylase inhibitors, a family of anticancer agents. J. Pharmacol. Exp. Ther. 2008, 325, 978–984. [Google Scholar]
- Dong, G.; Luo, J.; Kumar, V.; Dong, Z. Inhibitors of histone deacetylases suppress cisplatin-induced p53 activation and apoptosis in renal tubular cells. Am. J. Physiol. Renal Physiol. 2010, 298, F293–F300. [Google Scholar]
- Arany, I.; Herbert, J.; Herbert, Z.; Safirstein, R.L. Restoration of CREB function ameliorates cisplatin cytotoxicity in renal tubular cells. Am. J. Physiol. Renal Physiol. 2008, 294, F577–F581. [Google Scholar]
- Li, H.-F.; Cheng, C.-F.; Liao, W.-J.; Lin, H.; Yang, R.-B. ATF3-mediated epigenetic regulation protects against acute kidney injury. J. Am. Soc. Nephrol. 2010, 21, 1003–1013. [Google Scholar]
- Jo, S.K.; Cho, W.Y.; Sung, S.A.; Kim, H.K.; Won, N.H. MEK inhibitor, U0126, attenuates cisplatin-induced renal injury by decreasing inflammation and apoptosis. Kidney Int. 2005, 67, 458–466. [Google Scholar]
- Francescato, H.D.; Costa, R.S.; Junior, F.B.; Coimbra, T.M. Effect of JNK inhibition on cisplatin-induced renal damage. Nephrol. Dial. Transplant. 2007, 22, 2138–2148. [Google Scholar]
- Ramesh, G.; Reeves, W.B. p38 MAP kinase inhibition ameliorates cisplatin nephrotoxicity in mice. Am. J. Physiol. Renal Physiol. 2005, 289, F166–F174. [Google Scholar]
- Kim, Y.K.; Kim, H.J.; Kwon, C.H.; Kim, J.H.; Woo, J.S.; Jung, J.S.; Kim, J.M. Role of ERK activation in cisplatin-induced apoptosis in OK renal epithelial cells. J. Appl. Toxicol. 2005, 25, 374–382. [Google Scholar]
- Clark, J.S.; Faisal, A.; Baliga, R.; Nagamine, Y.; Arany, I. Cisplatin induces apoptosis through the ERK-p66shc pathway in renal proximal tubule cells. Cancer Lett. 2010, 297, 165–170. [Google Scholar]
- Baliga, R.; Zhang, Z.; Baliga, M.; Ueda, N.; Shah, S.V. In vitro and in vivo evidence suggesting a role for iron in cisplatin-induced nephrotoxicity. Kidney Int. 1998, 53, 394–401. [Google Scholar]
- Davis, C.; Nick, H.; Agarwal, A. Manganese superoxide dismutase attenuates cisplatin-induced renal injury: Importance of superoxide. J. Am. Soc. Nephrol. 2001, 12, 2683–2690. [Google Scholar]
- Appendroth, D.; Frob, S.; Kersten, L.; Splinter, F.K.; Winnefeld, K. Protective effects of Vitamin E and C on cisplatin nephrotoxicity in developing rats. Arch. Toxicol. 1997, 71, 677–683. [Google Scholar]
- Dickey, D.T.; Wu, Y.J.; Muldoon, L.L.; Neuwelt, E.A. Protection against cisplatin-induced toxicities by N-acetylcysteine and sodium thiosulfate as assessed at the molecular, cellular, and in vivo levels. J. Pharmacol. Exp. Ther. 2005, 314, 1052–1058. [Google Scholar]
- Ma, S.F.; Nishikawa, M.; Hyoudou, K.; Takahashi, R.; Ikemura, M.; Kobayashi, Y.; Yamashita, F.; Hashida, M. Combining cisplatin with cationized catalase decreases nephrotoxicity while improving antitumor activity. Kidney Int. 2007, 72, 1474–1482. [Google Scholar]
- Naziroglu, M.; Karaoglu, A.; Aksoy, A.O. Selenium and high dose vitamin E administration protects cisplatin-induced oxidative damage to renal, liver and lens tissues in rats. Toxicology 2004, 195, 221–230. [Google Scholar]
- Kelly, K.J.; Meehan, S.M.; Colvin, R.B.; Williams, W.W.; Bonventre, J.V. Protection from toxicant-mediated renal injury in the rat with anti-CD54 antibody. Kidney Int. 1999, 56, 922–931. [Google Scholar]
- Deng, J.; Kohda, Y.; Chiao, H.; Wang, Y.; Hu, X.; Hewitt, S.; Miyaja, T.; McLeroy, P.; Nibhanupudy, B.; Li, S.; Star, R. Interleukin-10 inhibits ischemic and cisplatin-induced acute renal injury. Kidney Int. 2001, 60, 2118–2128. [Google Scholar]
- Kim, Y.; Choi, T.; Kwon, C.; Kim, J.; Woo, J.; Jung, J. Beneficial effect of pentoxifylline on cisplatin-induced acute renal failure in rabbits. Ren. Fail. 2003, 25, 909–922. [Google Scholar]
- Zhang, B.; Ramesh, G.; Norbury, C.; Reeves, W.B. Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-α produced by renal parenchymal cells. Kidney Int. 2007, 72, 37–44. [Google Scholar]
- Barbara, J.A.J.; Smith, W.; Gamble, J.; Ostade, X.; Vandenabelle, P.; Tavernier, J.; Fiers, W.; Vadas, M.; Lopez, A. Dissociation of TNF-α cytotoxic and proinflammatory activities by p55 receptor- and p75 receptor-selective TNF-α mutants. EMBO J. 1994, 14, 843–850. [Google Scholar]
- Locksley, R.; Killeen, N.; Lenardo, M. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar]
- Faubel, S.; Lewis, E.C.; Reznikov, L.; Ljubanovic, D.; Hoke, T.S.; Somerset, H.; Oh, D.-J.; Lu, L.; Klein, C.L.; Dinarello, C.A.; Edelstein, C.L. Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1beta, IL-18, IL-6, and neutrophil infiltration in the kidney. J. Pharmacol. Exp. Ther. 2007, 322, 8–15. [Google Scholar]
- Lu, L.H.; Oh, D.J.; Dursun, B.; He, Z.; Hoke, T.S.; Faubel, S.; Edelstein, C.L. Increased macrophage infiltration and fractalkine expression in cisplatin-induced acute renal failure in mice. J. Pharmacol. Exp. Ther. 2008, 324, 111–117. [Google Scholar]
- Faubel, S.; Ljubanovic, D.; Reznikov, L.; Somerset, H.; Dinarello, C.A.; Edelstein, C.L. Caspase-1-deficient mice are protected against cisplatin-induced apoptosis and acute tubular necrosis. Kidney Int. 2004, 66, 2202–2213. [Google Scholar]
- Tadagavadi, R.K.; Reeves, W.B. Endogenous IL-10 attenuates cisplatin nephrotoxicity: Role of dendritic cells. J. Immunol. 2010, 185, 4904–4911. [Google Scholar]
- de Waal Malefyt, R. IL-10. In Cytokine Reference; Oppenheim, J.J., Feldmann, M., Eds.; Academic Press: San Diego, CA, USA, 2001; Volume 1, pp. 165–185. [Google Scholar]
- Zhang, B.; Ramesh, G.; Uematsu, S.; Akira, S.; Reeves, W.B. TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J. Am. Soc. Nephrol. 2008, 19, 923–932. [Google Scholar]
- Zhang, B.; Ramesh, G.; Wang, W.; Kwon, O.; Ahn, K.; Freeman, W.; Reeves, W.B. Urinary Cytokine Profiles in Different Forms of Acute Kidney Injury. 2010; manuscript in preparation. [Google Scholar]
- McDuffie, J.E.; Sablad, M.; Ma, J.; Snook, S. Urinary parameters predictive of cisplatin-induced acute renal injury in dogs. Cytokine 2010. [Google Scholar]
- Gluba, A.; Banach, M.; Hannam, S.; Mikhailidis, D.P.; Sakowicz, A.; Rysz, J. The role of Toll-like receptors in renal diseases. Nat. Rev. Nephrol. 2010, 6, 224–235. [Google Scholar]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar]
- Cunningham, P.N.; Dyanov, H.M.; Park, P.; Wang, J.; Newell, K.A.; Quigg, R.J. Acute renal failure in endotoxemia is caused by TNF acting directly on TNF receptor-1 in kidney. J. Immunol. 2002, 168, 5817–5823. [Google Scholar]
- Cunningham, P.N.; Wang, Y.; Guo, R.; He, G.; Quigg, R.J. Role of toll-like receptor 4 in endotoxin-induced acute renal failure. J. Immunol. 2004, 172, 2629–2635. [Google Scholar]
- Zager, R.A.; Johnson, A.C.M.; Hanson, S.Y.; Lund, S. Acute nephrotoxic and obstructive injury primes the kidney to endotoxin-driven cytokine/chemokine production. Kidney Int. 2006, 69, 1181–1188. [Google Scholar]
- Ramesh, G.; Kimball, S.R.; Jefferson, L.S.; Reeves, W.B. Endotoxin and cisplatin synergistically stimulate TNF-{alpha} production by renal epithelial cells. Am. J. Physiol. Renal Physiol. 2007, 292, F812–F819. [Google Scholar]
- Ramesh, G.; Zhang, B.; Uematsu, S.; Akira, S.; Reeves, W.B. Endotoxin and cisplatin synergistically induce renal dysfunction and cytokine production in mice. Am. J. Physiol. Renal Physiol. 2007, 293, F325–F332. [Google Scholar]
- Tsung, A.; Klune, J.R.; Zhang, X.; Jeyabalan, G.; Cao, Z.; Peng, X.; Stolz, D.B.; Geller, D.A.; Rosengart, M.R.; Billiar, T.R. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J. Exp. Med. 2007, 204, 2913–2923. [Google Scholar]
- Pan, P.; Cardinal, J.; Dhupar, R.; Rosengart, M.R.; Lotze, M.T.; Geller, D.A.; Billiar, T.R.; Tsung, A. Low-dose cisplatin administration in murine cecal ligation and puncture prevents the systemic release of HMGB1 and attenuates lethality. J. Leukoc. Biol. 2009, 86, 625–632. [Google Scholar]
- Leemans, J.C.; Stokman, G.; Claessen, N.; Rouschop, K.M.; Teske, G.J.D.; Kirschning, C.J.; Akira, S.; van der Poll, T.; Weening, J.J.; Florquin, S. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J. Clin. Invest. 2005, 115, 2894–2903. [Google Scholar]
- Zubin, M.B.; Vinod, P.B.; Lee, M.O.; Hebroon, O.; George, P.; Ronald, P.D. Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury. Hepatology 2010, 51, 621–632. [Google Scholar]
- Yi, A.-K.; Yoon, H.; Park, J.-E.; Kim, B.-S.; Kim, H.J.; Martinez-Hernandez, A. CpG DNA-mediated induction of acute liver injury in d-Galactosamine-sensitized mice: The mitochondrial apoptotic pathway-dependent death of hepatocytes. J. Biol. Chem. 2006, 281, 15001–15012. [Google Scholar]
- Yasuda, H.; Leelahavanichkul, A.; Tsunoda, S.; Dear, J.W.; Takahashi, Y.; Ito, S.; Hu, X.; Zhou, H.; Doi, K.; Childs, R.; Klinman, D.M.; Yuen, P.S.; Star, R.A. Chloroquine and inhibition of Toll-like receptor 9 protect from sepsis-induced acute kidney injury. Am. J. Physiol. Renal Physiol. 2008, 294, F1050–F1058. [Google Scholar]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar]
- Tadagavadi, R.K.; Reeves, W.B. TLR9 in cisplatin nephrotoxicity. 2010; College of Medicine, Pennsylvania State University, Hershey, PA, USA, unpublished work. [Google Scholar]
- Tadagavadi, R.K.; Reeves, W.B. Renal dendritic cells ameliorate nephrotoxic acute kidney injury. J. Am. Soc. Nephrol. 2010, 21, 53–63. [Google Scholar]
- Oh, D.-J.; Dursun, B.; He, Z.; Lu, L.; Hoke, T.S.; Ljubanovic, D.; Faubel, S.; Edelstein, C.L. Fractalkine receptor (CX3CR1) inhibition is protective against ischemic acute renal failure in mice. Am. J. Physiol. Renal Physiol. 2008, 294, F264–F271. [Google Scholar]
- Li, L.; Huang, L.; Sung, S.S.; Vergis, A.L.; Rosin, D.L.; Rose, C.E., Jr.; Lobo, P.I.; Okusa, M.D. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia-reperfusion injury. Kidney Int. 2008, 74, 1526–1537. [Google Scholar]
- Tarang, S.; Sodhi, A.; Chauhan, P. Differential expression of Toll-like receptors in murine peritoneal macrophages in vitro on treatment with cisplatin. Int. Immunol. 2007, 19, 635–643. [Google Scholar]
- Chauhan, P.; Sodhi, A.; Shrivastava, A. Cisplatin primes murine peritoneal macrophages for enhanced expression of nitric oxide, proinflammatory cytokines, TLRs, transcription factors and activation of MAP kinases upon co-incubation with L929 cells. Immunobiology 2009, 214, 197–209. [Google Scholar]
- Burne, M.; Daniels, F.; Ghandourm, A.; Mauiyyedi, S.; Colvin, R.; O’Donnell, M.; Rabb, H. Identification of the CD4+ T Cells as a major pathogenic factor in ischemic acute renal failure. J. Clin. Invest. 2001, 108, 1283–1290. [Google Scholar]
- Liu, M.; Chien, C.-C.; Burne-Taney, M.; Molls, R.R.; Racusen, L.C.; Colvin, R.B.; Rabb, H. A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity. J. Am. Soc. Nephrol. 2006, 17, 765–774. [Google Scholar]
- Lee, V.W.; Wang, Y.M.; Wang, Y.P.; Zheng, D.; Polhill, T.; Cao, Q.; Wu, H.; Alexander, I.E.; Alexander, S.I.; Harris, D.C. Regulatory immune cells in kidney disease. Am. J. Physiol. Renal Physiol. 2008, 295, F335–F342. [Google Scholar]
- Gandolfo, M.T.; Jang, H.R.; Bagnasco, S.; Ko, G.-J.; Agreda, P.; Satpute, S.; Crow, M.T.; King, L.S.; Rabb, H. Foxp3+ regulatory T cells participate in repair of ischemic acute kidney injury. Kidney Int. 2009, 76, 717–729. [Google Scholar]
- Kinsey, G.R.; Sharma, R.; Huang, L.; Li, L.; Vergis, A.L.; Ye, H.; Ju, S.-T.; Okusa, M.D. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2009, 20, 1744–1753. [Google Scholar]
- Lee, H.; Nho, D.; Chung, H.S.; Lee, H.; Shin, M.K.; Kim, S.H.; Bae, H. CD4(+)CD25(+) regulatory T cells attenuate cisplatin-induced nephrotoxicity in mice. Kidney Int. 2010. [Google Scholar]
- Laouar, Y.; Town, T.; Jeng, D.; Tran, E.; Wan, Y.; Kuchroo, V.K.; Flavell, R.A. TGF-beta signaling in dendritic cells is a prerequisite for the control of autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2008, 105, 10865–10870. [Google Scholar]
- Akbari, O.; DeKruyff, R.H.; Umetsu, D.T. Pulmonary dendritic cells producing IL-10 mediate tolerance induced by respiratory exposure to antigen. Nat. Immunol. 2001, 2, 725–731. [Google Scholar]
- Akbari, O.; Freeman, G.J.; Meyer, E.H.; Greenfield, E.A.; Chang, T.T.; Sharpe, A.H.; Berry, G.; DeKruyff, R.H.; Umetsu, D.T. Antigen-specific regulatory T cells develop via the ICOS-ICOS-ligand pathway and inhibit allergen-induced airway hyperreactivity. Nat. Med. 2002, 8, 1024–1032. [Google Scholar]
- Jung, S.; Unutmaz, D.; Wong, P.; Sano, G.; De los Santos, K.; Sparwasser, T.; Wu, S.; Vuthoori, S.; Ko, K.; Zavala, F.; Pamer, E.G.; Littman, D.R.; Lang, R.A. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 2002, 17, 211–220. [Google Scholar]
- van Rijt, L.S.; Jung, S.; Kleinjan, A.; Vos, N.; Willart, M.; Duez, C.; Hoogsteden, H.C.; Lambrecht, B.N. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J. Exp. Med. 2005, 201, 981–991. [Google Scholar]
- Soos, T.J.; Sims, T.N.; Barisoni, L.; Lin, K.; Littman, D.R.; Dustin, M.L.; Nelson, P.J. CX3CR1+ interstitial dendritic cells form a contiguous network throughout the entire kidney. Kidney Int. 2006, 70, 591–596. [Google Scholar]
- Dong, X.; Swaminathan, S.; Bachman, L.A.; Croatt, A.J.; Nath, K.A.; Griffin, M.D. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int. 2007, 71, 619–628. [Google Scholar]
- Cornelison, T.L.; Reed, E. Nephrotoxicity and hydration management for cisplatin, carboplatin, and ormaplatin. Gynecol. Oncol. 1993, 50, 147–158. [Google Scholar]
- Lehane, D.; Winston, A.; Gray, R.; Daskal, Y. The effect of diuretic pre-treatment on clinical, morphological and ultrastructural cis-platinum induced nephrotoxicity. Int. J. Radiat. Oncol. Biol. Phys. 1979, 5, 1393–1399. [Google Scholar]
- Al-Sarraf, M.; Fletcher, W.; Oishi, N.; Pugh, R.; Hewlett, J.S.; Balducci, L.; McCracken, J.; Padilla, F. Cisplatin hydration with and without mannitol diuresis in refractory disseminated malignant melanoma: A southwest oncology group study. Cancer Treat. Rep. 1982, 66, 31–35. [Google Scholar]
- Levi, J.; Jacobs, C.; Kalman, S.M.; McTigue, M.; Weiner, M.W. Mechanism of cis-platinum nephrotoxicity: I. Effects of sulfhydryl groups in rat kidneys. J. Pharmacol. Exp. Ther. 1980, 213, 545–550. [Google Scholar] [PubMed]
- Safirstein, R.; Winston, J.; Moel, D.; Dikman, S.; Guttenplan, J. Cisplatin nephrotoxicity: Insights into mechanism. Int. J. Androl. 1987, 10, 325–346. [Google Scholar]
- Hensley, M.L.; Hagerty, K.L.; Kewalramani, T.; Green, D.M.; Meropol, N.J.; Wasserman, T.H.; Cohen, G.I.; Emami, B.; Gradishar, W.J.; Mitchell, R.B.; et al. American Society of Clinical Oncology 2008 clinical practice guideline update: Use of chemotherapy and radiation therapy protectants. J. Clin. Oncol. 2009, 27, 127–145. [Google Scholar]
- Castiglione, F.; Mola, A.D.; Porcile, G. Protection of normal tissues from radiation and cytotoxic therapy: The development of amifostine. Tumori 1999, 85, 85–91. [Google Scholar]
- Capizzi, R.L. Amifostine reduces the incidence of cumulative nephrotoxicity from cisplatin: Laboratory and clinical aspects. Semin. Oncol. 1999, 26, 72–81. [Google Scholar]
- Yasuo, M.; Yasuhiro, M.; Tetsuya, H.; Nobuhiro, N.; Kazunori, K.; Takanori, K.; William, J.M.H.; Fuminori, M.; Tadao, K. Cisplatin-incorporated polymeric micelles eliminate nephrotoxicity, while maintaining antitumor activity. Cancer Sci. 2001, 92, 328–336. [Google Scholar]
- Uchino, H.; Matsumura, Y.; Negishi, T.; Koizumi, F.; Hayashi, T.; Honda, T.; Nishiyama, N.; Kataoka, K.; Naito, S.; Kakizoe, T. Cisplatin-incorporating polymeric micelles (NC-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br. J. Cancer 2005, 93, 678–687. [Google Scholar]
- Franke, R.M.; Kosloske, A.M.; Lancaster, C.S.; Filipski, K.; Hu, C.; Zolk, O.; Mathijssen, R.H.; Sparreboom, A. Influence of Oct1/Oct2-deficiency on cisplatin-induced changes in urinary N-Acetyl-{beta}-D-Glucosaminidase. Clin. Cancer Res. 2010, 16, 4198–4206. [Google Scholar]
- Sleijfer, D.T.; Offerman, J.J.G.; Mulder, N.H.; Verweij, M.; van der Hem, G.K.; Schraffordt Koops, H.; Meijer, S. The protective potential of the combination of verapamil and cimetidine on cisplatin-induced nephrotoxicity in man. Cancer 1987, 60, 2823–2828. [Google Scholar]
- Hanigan, M.H.; Gallagher, B.C.; Taylor, P.T., Jr.; Large, M.K. Inhibition of gamma-glutamyl transpeptidase activity by acivicin in vivo protects the kidney from cisplatin-induced toxicity. Cancer Res. 1994, 54, 5925–5929. [Google Scholar]
- Hausheer, F.H.; Kochat, H.; Parker, A.R.; Ding, D.; Yao, S.; Hamilton, S.E.; Petluru, P.N.; Leverett, B.D.; Bain, S.H.; Saxe, J.D. New approaches to drug discovery and development: A mechanism-based approach to pharmaceutical research and its application to BNP7787, a novel chemoprotective agent. Cancer Chemother. Pharmacol. 2003, 52, 3–15. [Google Scholar]
- Wu, Y.J.; Muldoon, L.L.; Neuwelt, E.A. The chemoprotective agent N-Acetylcysteine blocks cisplatin-induced apoptosis through caspase signaling pathway. J. Pharmacol. Exp. Ther. 2005, 312, 424–431. [Google Scholar]
- Lynch, E.D.; Gu, R.; Pierce, C.; Kil, J. Reduction of acute cisplatin ototoxicity and nephrotoxicity in rats by oral administration of allopurinol and ebselen. Hear. Res. 2005, 201, 81–89. [Google Scholar]
- Karimi, G.; Ramezani, M.; Tahoonian, Z. Cisplatin nephrotoxicity and protection by milk thistle extract in rats. Evid. Based Complement. Alternat. Med. 2005, 2, 383–386. [Google Scholar]
- Pan, H.; Mukhopadhyay, P.; Rajesh, M.; Patel, V.; Mukhopadhyay, B.; Gao, B.; Haskó, G.; Pacher, P. Cannabidiol attenuates cisplatin-induced nephrotoxicity by decreasing oxidative/nitrosative stress, inflammation, and cell death. J. Pharmacol. Exp. Ther. 2009, 328, 708–714. [Google Scholar]
- Atessahin, A.; Yilmaz, S.; Karahan, I.; Ceribasi, A.O.; Karaoglu, A. Effects of lycopene against cisplatin-induced nephrotoxicity and oxidative stress in rats. Toxicology 2005, 212, 116–123. [Google Scholar]
- Li, S.; Gokden, N.; Okusa, M.D.; Bhatt, R.; Portilla, D. Anti-inflammatory effect of fibrate protects from cisplatin-induced ARF. Am. J. Physiol. Renal Physiol. 2005, 289, F469–F480. [Google Scholar]
- Tikoo, K.; Kumar, P.; Gupta, J. Rosiglitazone synergizes anticancer activity of cisplatin and reduces its nephrotoxicity in 7,12-dimethyl benz{a}anthracene (DMBA) induced breast cancer rats. BMC Cancer 2009, 9, 107. [Google Scholar]
- Kang, K.P.; Kim, D.H.; Jung, Y.J.; Lee, A.S.; Lee, S.; Lee, S.Y.; Jang, K.Y.; Sung, M.J.; Park, S.K.; Kim, W. Alpha-lipoic acid attenuates cisplatin-induced acute kidney injury in mice by suppressing renal inflammation. Nephrol. Dial. Transplant. 2009, 24, 3012–3020. [Google Scholar]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar]
- Kulbe, H.; Thompson, R.; Wilson, J.L.; Robinson, S.; Hagemann, T.; Fatah, R.; Gould, D.; Ayhan, A.; Balkwill, F. The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res. 2007, 67, 585–592. [Google Scholar]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Invest. 2008, 118, 560–570. [Google Scholar]
- Waterson, A.; Bower, M. TNF and cancer: Good or bad? Cancer Ther. 2004, 2, 131–148. [Google Scholar]
- Chen, R.; Alvero, A.B.; Silasi, D.A.; Steffensen, K.D.; Mor, G. Cancers take their Toll-the function and regulation of Toll-like receptors in cancer cells. Oncogene 2008, 27, 225–233. [Google Scholar]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin Nephrotoxicity. Toxins 2010, 2, 2490-2518. https://doi.org/10.3390/toxins2112490
Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of Cisplatin Nephrotoxicity. Toxins. 2010; 2(11):2490-2518. https://doi.org/10.3390/toxins2112490
Chicago/Turabian StyleMiller, Ronald P., Raghu K. Tadagavadi, Ganesan Ramesh, and William Brian Reeves. 2010. "Mechanisms of Cisplatin Nephrotoxicity" Toxins 2, no. 11: 2490-2518. https://doi.org/10.3390/toxins2112490
APA StyleMiller, R. P., Tadagavadi, R. K., Ramesh, G., & Reeves, W. B. (2010). Mechanisms of Cisplatin Nephrotoxicity. Toxins, 2(11), 2490-2518. https://doi.org/10.3390/toxins2112490