Clostridial Neurotoxins: Mechanism of SNARE Cleavage and Outlook on Potential Substrate Specificity Reengineering

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Cleavage of SNAREs

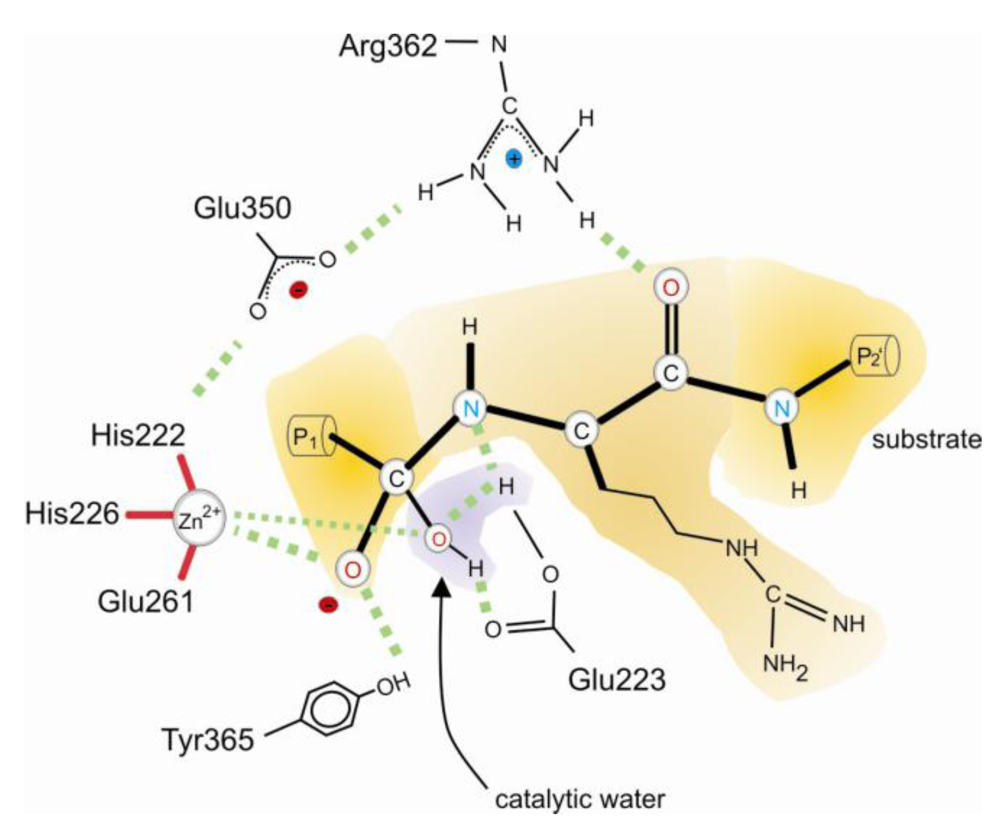
3. Catalytic Mechanism

4. Mode of Substrate Recognition

5. Reengineered CNT Catalytic Domains to Target Non-substrate SNAREs

Acknowledgements
References
- Dolly, J.O.; Black, J.; Williams, R.S.; Melling, J. Acceptors for botulinum neurotoxin reside on motor nerve terminals and mediate its internalization. Nature 1984, 307, 457–460. [Google Scholar]
- Dong, M.; Richards, D.A.; Goodnough, M.C.; Tepp, W.H.; Johnson, E.A.; Chapman, E.R. Synaptotagmins I and II mediate entry of botulinum neurotoxin B into cells. J. Cell Biol. 2003, 162, 1293–1303. [Google Scholar]
- Dong, M.; Tepp, W.H.; Liu, H.; Johnson, E.A.; Chapman, E.R. Mechanism of botulinum neurotoxin B and G entry into hippocampal neurons. J. Cell Biol. 2007, 179, 1511–1522. [Google Scholar]
- Nishiki, T.; Kamata, Y.; Nemoto, Y.; Omori, A.; Ito, T.; Takahashi, M.; Kozaki, S. Identification of protein receptor for Clostridium botulinum type B neurotoxin in rat brain synaptosomes. J. Biol. Chem. 1994, 269, 10498–10503. [Google Scholar]
- Rummel, A.; Karnath, T.; Henke, T.; Bigalke, H.; Binz, T. Synaptotagmins I and II act as nerve cell receptors for botulinum neurotoxin G. J. Biol. Chem. 2004, 279, 30865–30870. [Google Scholar]
- Dong, M.; Liu, H.; Tepp, W.H.; Johnson, E.A.; Janz, R.; Chapman, E.R. Glycosylated SV2A and SV2B mediate the entry of botulinum neurotoxin E into neurons. Mol. Biol. Cell 2008, 19, 5226–5237. [Google Scholar]
- Dong, M.; Yeh, F.; Tepp, W.H.; Dean, C.; Johnson, E.A.; Janz, R.; Chapman, E.R. SV2 is the protein receptor for botulinum neurotoxin A. Science 2006, 312, 592–596. [Google Scholar]
- Mahrhold, S.; Rummel, A.; Bigalke, H.; Davletov, B.; Binz, T. The synaptic vesicle protein 2C mediates the uptake of botulinum neurotoxin A into phrenic nerves. FEBS Lett. 2006, 580, 2011–2014. [Google Scholar]
- Fu, Z.; Chen, C.; Barbieri, J.T.; Kim, J.J.; Baldwin, M.R. Glycosylated SV2 and gangliosides as dual receptors for botulinum neurotoxin serotype F. Biochemistry 2009, 48, 5631–5641. [Google Scholar]
- Rummel, A.; Hafner, K.; Mahrhold, S.; Darashchonak, N.; Holt, M.; Jahn, R.; Beermann, S.; Karnath, T.; Bigalke, H.; Binz, T. Botulinum neurotoxins C, E and F bind gangliosides via a conserved binding site prior to stimulation-dependent uptake with botulinum neurotoxin F utilising the three isoforms of SV2 as second receptor. J. Neurochem. 2009, 110, 1942–1954. [Google Scholar]
- Muraro, L.; Tosatto, S.; Motterlini, L.; Rossetto, O.; Montecucco, C. The N-terminal half of the receptor domain of botulinum neurotoxin A binds to microdomains of the plasma membrane. Biochem. Biophys. Res. Commun. 2009, 380, 76–80. [Google Scholar]
- Fischer, A.; Montal, M. Single molecule detection of intermediates during botulinum neurotoxin translocation across membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 10447–10452. [Google Scholar]
- Koriazova, L.K.; Montal, M. Translocation of botulinum neurotoxin light chain protease through the heavy chain channel. Nat. Struct. Biol. 2003, 10, 13–18. [Google Scholar]
- Schiavo, G.; Matteoli, M.; Montecucco, C. Neurotoxins affecting neuroexocytosis. Physiol. Rev. 2000, 80, 717–766. [Google Scholar]
- Gill, D.M. Bacterial toxins: a table of lethal amounts. Microbiol. Rev. 1982, 46, 86–94. [Google Scholar]
- Arnon, S.S.; Schechter, R.; Inglesby, T.V.; Henderson, D.A.; Bartlett, J.G.; Ascher, M.S.; Eitzen, E.; Fine, A.D.; Hauer, J.; Layton, M.; Lillibridge, S.; Osterholm, M.T.; O'Toole, T.; Parker, G.; Perl, T.M.; Russell, P.K.; Swerdlow, D.L.; Tonat, K. Botulinum toxin as a biological weapon: Medical and public health management. Jama 2001, 285, 1059–1070. [Google Scholar]
- Bigalke, H.; Rummel, A. Medical aspects of toxin weapons. Toxicology 2005, 214, 210–220. [Google Scholar]
- Davletov, B.; Bajohrs, M.; Binz, T. Beyond BOTOX: Advantages and limitations of individual botulinum neurotoxins. Trends Neurosci. 2005, 28, 446–452. [Google Scholar]
- Jongeneel, C.V.; Bouvier, J.; Bairoch, A. A unique signature identifies a family of zinc-dependent metallopeptidases. FEBS Lett. 1989, 242, 211–214. [Google Scholar]
- Binz, T.; Kurazono, H.; Wille, M.; Frevert, J.; Wernars, K.; Niemann, H. The complete sequence of botulinum neurotoxin type A and comparison with other clostridial neurotoxins. J. Biol. Chem. 1990, 265, 9153–9158. [Google Scholar]
- Thompson, D.E.; Brehm, J.K.; Oultram, J.D.; Swinfield, T.J.; Shone, C.C.; Atkinson, T.; Melling, J.; Minton, N.P. The complete amino acid sequence of the Clostridium botulinum type A neurotoxin, deduced by nucleotide sequence analysis of the encoding gene. Eur. J. Biochem. 1990, 189, 73–81. [Google Scholar]
- Schiavo, G.; Rossetto, O.; Santucci, A.; DasGupta, B.R.; Montecucco, C. Botulinum neurotoxins are zinc proteins. J. Biol. Chem. 1992, 267, 23479–23483. [Google Scholar]
- Schiavo, G.; Poulain, B.; Rossetto, O.; Benfenati, F.; Tauc, L.; Montecucco, C. Tetanus toxin is a zinc protein and its inhibition of neurotransmitter release and protease activity depend on zinc. EMBO J. 1992, 11, 3577–3583. [Google Scholar]
- Baumert, M.; Maycox, P.R.; Navone, F.; De Camilli, P.; Jahn, R. Synaptobrevin: an integral membrane protein of 18,000 daltons present in small synaptic vesicles of rat brain. EMBO J. 1989, 8, 379–384. [Google Scholar]
- Schiavo, G.; Benfenati, F.; Poulain, B.; Rossetto, O.; Polverino de Laureto, P.; DasGupta, B.R.; Montecucco, C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 1992, 359, 832–835. [Google Scholar]
- Trimble, W.S.; Cowan, D.M.; Scheller, R.H. VAMP-1: A synaptic vesicle-associated integral membrane protein. Proc. Natl. Acad. Sci. USA 1988, 85, 4538–4542. [Google Scholar]
- Schiavo, G.; Rossetto, O.; Catsicas, S.; Polverino de Laureto, P.; DasGupta, B.R.; Benfenati, F.; Montecucco, C. Identification of the nerve terminal targets of botulinum neurotoxin serotypes A, D, and E. J. Biol. Chem. 1993, 268, 23784–23787. [Google Scholar]
- Schiavo, G.; Shone, C.C.; Rossetto, O.; Alexander, F.C.; Montecucco, C. Botulinum neurotoxin serotype F is a zinc endopeptidase specific for VAMP/synaptobrevin. J. Biol. Chem. 1993, 268, 11516–11519. [Google Scholar]
- Yamasaki, S.; Binz, T.; Hayashi, T.; Szabo, E.; Yamasaki, N.; Eklund, M.; Jahn, R.; Niemann, H. Botulinum neurotoxin type G proteolyses the Ala81-Ala82 bond of rat synaptobrevin 2. Biochem. Biophys. Res. Commun. 1994, 200, 829–835. [Google Scholar]
- Blasi, J.; Chapman, E.R.; Link, E.; Binz, T.; Yamasaki, S.; De Camilli, P.; Sudhof, T.C.; Niemann, H.; Jahn, R. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature 1993, 365, 160–163. [Google Scholar]
- Foran, P.; Lawrence, G.W.; Shone, C.C.; Foster, K.A.; Dolly, J.O. Botulinum neurotoxin C1 cleaves both syntaxin and SNAP-25 in intact and permeabilized chromaffin cells: correlation with its blockade of catecholamine release. Biochemistry 1996, 35, 2630–2636. [Google Scholar]
- Osen-Sand, A.; Staple, J.K.; Naldi, E.; Schiavo, G.; Rossetto, O.; Petitpierre, S.; Malgaroli, A.; Montecucco, C.; Catsicas, S. Common and distinct fusion proteins in axonal growth and transmitter release. J. Comp. Neurol. 1996, 367, 222–234. [Google Scholar]
- Williamson, L.C.; Halpern, J.L.; Montecucco, C.; Brown, J.E.; Neale, E.A. Clostridial neurotoxins and substrate proteolysis in intact neurons: botulinum neurotoxin C acts on synaptosomal-associated protein of 25 kDa. J. Biol. Chem. 1996, 271, 7694–7699. [Google Scholar]
- Binz, T.; Blasi, J.; Yamasaki, S.; Baumeister, A.; Link, E.; Sudhof, T.C.; Jahn, R.; Niemann, H. Proteolysis of SNAP-25 by types E and A botulinal neurotoxins. J. Biol. Chem. 1994, 269, 1617–1620. [Google Scholar]
- Schiavo, G.; Santucci, A.; Dasgupta, B.R.; Mehta, P.P.; Jontes, J.; Benfenati, F.; Wilson, M.C.; Montecucco, C. Botulinum neurotoxins serotypes A and E cleave SNAP-25 at distinct COOH-terminal peptide bonds. FEBS Lett. 1993, 335, 99–103. [Google Scholar]
- Vaidyanathan, V.V.; Yoshino, K.; Jahnz, M.; Dorries, C.; Bade, S.; Nauenburg, S.; Niemann, H.; Binz, T. Proteolysis of SNAP-25 isoforms by botulinum neurotoxin types A, C, and E: Domains and amino acid residues controlling the formation of enzyme-substrate complexes and cleavage. J. Neurochem. 1999, 72, 327–337. [Google Scholar]
- Blasi, J.; Chapman, E.R.; Yamasaki, S.; Binz, T.; Niemann, H.; Jahn, R. Botulinum neurotoxin C1 blocks neurotransmitter release by means of cleaving HPC-1/syntaxin. EMBO J. 1993, 12, 4821–4828. [Google Scholar]
- Bennett, M.K.; Calakos, N.; Scheller, R.H. Syntaxin: A synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science 1992, 257, 255–259. [Google Scholar]
- Inoue, A.; Obata, K.; Akagawa, K. Cloning and sequence analysis of cDNA for a neuronal cell membrane antigen, HPC-1. J. Biol. Chem. 1992, 267, 10613–10619. [Google Scholar]
- Schiavo, G.; Shone, C.C.; Bennett, M.K.; Scheller, R.H.; Montecucco, C. Botulinum neurotoxin type C cleaves a single Lys-Ala bond within the carboxyl-terminal region of syntaxins. J. Biol. Chem. 1995, 270, 10566–10570. [Google Scholar]
- Sutton, R.B.; Fasshauer, D.; Jahn, R.; Brunger, A.T. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2. 4 A resolution. Nature 1998, 395, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Jahn, R.; Scheller, R.H. SNAREs--engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 2006, 7, 631–643. [Google Scholar]
- Humeau, Y.; Doussau, F.; Grant, N.J.; Poulain, B. How botulinum and tetanus neurotoxins block neurotransmitter release. Biochimie 2000, 82, 427–446. [Google Scholar]
- Sikorra, S.; Henke, T.; Galli, T.; Binz, T. Substrate recognition mechanism of VAMP/synaptobrevin-cleaving clostridial neurotoxins. J. Biol. Chem. 2008, 283, 21145–21152. [Google Scholar]
- Sikorra, S.; Henke, T.; Swaminathan, S.; Galli, T.; Binz, T. Identification of the amino acid residues rendering TI-VAMP insensitive toward botulinum neurotoxin B. J. Mol. Biol. 2006, 357, 574–582. [Google Scholar]
- Hooper, N.M. Families of zinc metalloproteases. FEBS Lett. 1994, 354, 1–6. [Google Scholar]
- Rawlings, N.D.; Barrett, A.J. Evolutionary families of metallopeptidases. Methods Enzymol. 1995, 248, 183–228. [Google Scholar]
- Lacy, D.B.; Tepp, W.; Cohen, A.C.; DasGupta, B.R.; Stevens, R.C. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat. Struct. Biol. 1998, 5, 898–902. [Google Scholar]
- Breidenbach, M.A.; Brunger, A.T. 2.3 A crystal structure of tetanus neurotoxin light chain. Biochemistry 2005, 44, 7450–7457. [Google Scholar]
- Breidenbach, M.A.; Brunger, A.T. Substrate recognition strategy for botulinum neurotoxin serotype A. Nature 2004, 432, 925–929. [Google Scholar]
- Hangauer, D.G.; Monzingo, A.F.; Matthews, B.W. An interactive computer graphics study of thermolysin-catalyzed peptide cleavage and inhibition by N-carboxymethyl dipeptides. Biochemistry 1984, 23, 5730–5741. [Google Scholar]
- Matthews, B.W. Structural basis of the action of thermolysin and related zinc peptidases. Accounts Chem. Res. 1988, 21, 333–340. [Google Scholar]
- Li, L.; Binz, T.; Niemann, H.; Singh, B.R. Probing the mechanistic role of glutamate residue in the zinc-binding motif of type A botulinum neurotoxin light chain. Biochemistry 2000, 39, 2399–2405. [Google Scholar]
- Agarwal, R.; Eswaramoorthy, S.; Kumaran, D.; Binz, T.; Swaminathan, S. Structural analysis of botulinum neurotoxin type E catalytic domain and its mutant Glu212-->Gln reveals the pivotal role of the Glu212 carboxylate in the catalytic pathway. Biochemistry 2004, 43, 6637–6644. [Google Scholar]
- Agarwal, R.; Swaminathan, S. SNAP-25 substrate peptide (residues 180-183) binds to but bypasses cleavage by catalytically active Clostridium botulinum neurotoxin E. J. Biol. Chem. 2008, 283, 25944–25951. [Google Scholar]
- Kumaran, D.; Rawat, R.; Ahmed, S.A.; Swaminathan, S. Substrate binding mode and its implication on drug design for botulinum neurotoxin A. PLoS Pathogens 2008, 4, e1000165. [Google Scholar]
- Binz, T.; Bade, S.; Rummel, A.; Kollewe, A.; Alves, J. Arg(362) and Tyr(365) of the botulinum neurotoxin type a light chain are involved in transition state stabilization. Biochemistry 2002, 41, 1717–1723. [Google Scholar]
- Rossetto, O.; Caccin, P.; Rigoni, M.; Tonello, F.; Bortoletto, N.; Stevens, R.C.; Montecucco, C. Active-site mutagenesis of tetanus neurotoxin implicates TYR-375 and GLU-271 in metalloproteolytic activity. Toxicon 2001, 39, 1151–1159. [Google Scholar]
- Ahmed, S.A.; Olson, M.A.; Ludivico, M.L.; Gilsdorf, J.; Smith, L.A. Identification of residues surrounding the active site of type A botulinum neurotoxin important for substrate recognition and catalytic activity. Protein J. 2008, 27, 151–162. [Google Scholar]
- Silvaggi, N.R.; Wilson, D.; Tzipori, S.; Allen, K.N. Catalytic features of the botulinum neurotoxin A light chain revealed by high resolution structure of an inhibitory peptide complex. Biochemistry 2008, 47, 5736–5745. [Google Scholar]
- Agarwal, R.; Binz, T.; Swaminathan, S. Analysis of active site residues of botulinum neurotoxin E by mutational, functional, and structural studies: Glu335Gln is an apoenzyme. Biochemistry 2005, 44, 8291–8302. [Google Scholar]
- Jin, R.; Sikorra, S.; Stegmann, C.M.; Pich, A.; Binz, T.; Brunger, A.T. Structural and biochemical studies of botulinum neurotoxin serotype C1 light chain protease: Implications for dual substrate specificity. Biochemistry 2007, 46, 10685–10693. [Google Scholar]
- Cornille, F.; Martin, L.; Lenoir, C.; Cussac, D.; Roques, B.P.; Fournie-Zaluski, M.C. Cooperative exosite-dependent cleavage of synaptobrevin by tetanus toxin light chain. J. Biol. Chem. 1997, 272, 3459–3464. [Google Scholar]
- Foran, P.; Shone, C.C.; Dolly, J.O. Differences in the protease activities of tetanus and botulinum B toxins revealed by the cleavage of vesicle-associated membrane protein and various sized fragments. Biochemistry 1994, 33, 15365–15374. [Google Scholar]
- Schmidt, J.J.; Stafford, R.G. Botulinum neurotoxin serotype F: identification of substrate recognition requirements and development of inhibitors with low nanomolar affinity. Biochemistry 2005, 44, 4067–4073. [Google Scholar]
- Shone, C.C.; Quinn, C.P.; Wait, R.; Hallis, B.; Fooks, S.G.; Hambleton, P. Proteolytic cleavage of synthetic fragments of vesicle-associated membrane protein, isoform-2 by botulinum type B neurotoxin. Eur. J. Biochem. 1993, 217, 965–971. [Google Scholar]
- Yamasaki, S.; Baumeister, A.; Binz, T.; Blasi, J.; Link, E.; Cornille, F.; Roques, B.; Fykse, E.M.; Sudhof, T.C.; Jahn, R.; et al. Cleavage of members of the synaptobrevin/VAMP family by types D and F botulinal neurotoxins and tetanus toxin. J. Biol. Chem. 1994, 269, 12764–12772. [Google Scholar]
- Washbourne, P.; Pellizzari, R.; Baldini, G.; Wilson, M.C.; Montecucco, C. Botulinum neurotoxin types A and E require the SNARE motif in SNAP-25 for proteolysis. FEBS Lett. 1997, 418, 1–5. [Google Scholar]
- Pellizzari, R.; Rossetto, O.; Lozzi, L.; Giovedi, S.; Johnson, E.; Shone, C.C.; Montecucco, C. Structural determinants of the specificity for synaptic vesicle-associated membrane protein/synaptobrevin of tetanus and botulinum type B and G neurotoxins. J. Biol. Chem. 1996, 271, 20353–20358. [Google Scholar]
- Pellizzari, R.; Mason, S.; Shone, C.C.; Montecucco, C. The interaction of synaptic vesicle-associated membrane protein/synaptobrevin with botulinum neurotoxins D and F. FEBS Lett. 1997, 409, 339–342. [Google Scholar]
- Evans, E.R.; Sutton, J.M.; Gravett, A.; Shone, C.C. Analysis of the substrate recognition domain determinants of Botulinum Type B toxin using Phage display. Toxicon 2005, 46, 446–453. [Google Scholar]
- Wictome, M.; Rossetto, O.; Montecucco, C.; Shone, C.C. Substrate residues N-terminal to the cleavage site of botulinum type B neurotoxin play a role in determining the specificity of its endopeptidase activity. FEBS Lett. 1996, 386, 133–136. [Google Scholar]
- Chen, S.; Barbieri, J.T. Unique substrate recognition by botulinum neurotoxins serotypes A and E. J. Biol. Chem. 2006, 281, 10906–10911. [Google Scholar]
- Fu, Z.; Chen, S.; Baldwin, M.R.; Boldt, G.E.; Crawford, A.; Janda, K.D.; Barbieri, J.T.; Kim, J.J. Light chain of botulinum neurotoxin serotype A: Structural resolution of a catalytic intermediate. Biochemistry 2006, 45, 8903–8911. [Google Scholar]
- Silvaggi, N.R.; Boldt, G.E.; Hixon, M.S.; Kennedy, J.P.; Tzipori, S.; Janda, K.D.; Allen, K.N. Structures of clostridium botulinum neurotoxin serotype a light chain complexed with small-molecule inhibitors highlight active-site flexibility. Chem. Biol. 2007, 14, 533–542. [Google Scholar]
- Burnett, J.C.; Ruthel, G.; Stegmann, C.M.; Panchal, R.G.; Nguyen, T.L.; Hermone, A.R.; Stafford, R.G.; Lane, D.J.; Kenny, T.A.; McGrath, C.F.; Wipf, P.; Stahl, A.M.; Schmidt, J.J.; Gussio, R.; Brunger, A.T.; Bavari, S. Inhibition of metalloprotease botulinum serotype A from a pseudo-peptide binding mode to a small molecule that is active in primary neurons. J. Biol. Chem. 2007, 282, 5004–5014. [Google Scholar]
- Kumaran, D.; Rawat, R.; Ludivico, M.L.; Ahmed, S.A.; Swaminathan, S. Structure- and substrate-based inhibitor design for Clostridium botulinum neurotoxin serotype A. J. Biol. Chem. 2008, 283, 18883–18891. [Google Scholar]
- Zuniga, J.E.; Schmidt, J.J.; Fenn, T.; Burnett, J.C.; Arac, D.; Gussio, R.; Stafford, R.G.; Badie, S.S.; Bavari, S.; Brunger, A.T. A potent peptidomimetic inhibitor of botulinum neurotoxin serotype A has a very different conformation than SNAP-25 substrate. Structure 2008, 16, 1588–1597. [Google Scholar]
- Chen, S.; Kim, J.J.; Barbieri, J.T. Mechanism of substrate recognition by botulinum neurotoxin serotype A. J. Biol. Chem. 2007, 282, 9621–9627. [Google Scholar]
- Chen, S.; Barbieri, J.T. Multi-pocket recognition of SNAP25 by botulinum neurotoxin serotype E. J. Biol. Chem. 2007, 282, 25540–25547. [Google Scholar]
- Agarwal, R.; Schmidt, J.J.; Stafford, R.G.; Swaminathan, S. Mode of VAMP substrate recognition and inhibition of Clostridium botulinum neurotoxin F. Nat. Struct. Mol. Biol. 2009, 16, 789–794. [Google Scholar]
- Chaddock, J.A.; Purkiss, J.R.; Alexander, F.C.; Doward, S.; Fooks, S.J.; Friis, L.M.; Hall, Y.H.; Kirby, E.R.; Leeds, N.; Moulsdale, H.J.; Dickenson, A.; Green, G.M.; Rahman, W.; Suzuki, R.; Duggan, M.J.; Quinn, C.P.; Shone, C.C.; Foster, K.A. Retargeted clostridial endopeptidases: Inhibition of nociceptive neurotransmitter release in vitro, and antinociceptive activity in in vivo models of pain. Mov. Disord. 2004, 19, S42–S47. [Google Scholar]
- Duggan, M.J.; Quinn, C.P.; Chaddock, J.A.; Purkiss, J.R.; Alexander, F.C.; Doward, S.; Fooks, S.J.; Friis, L.M.; Hall, Y.H.; Kirby, E.R.; Leeds, N.; Moulsdale, H.J.; Dickenson, A.; Green, G.M.; Rahman, W.; Suzuki, R.; Shone, C.C.; Foster, K.A. Inhibition of release of neurotransmitters from rat dorsal root ganglia by a novel conjugate of a Clostridium botulinum toxin A endopeptidase fragment and Erythrina cristagalli lectin. J. Biol. Chem. 2002, 277, 34846–34852. [Google Scholar]
- Foster, K.A.; Adams, E.J.; Durose, L.; Cruttwell, C.J.; Marks, E.; Shone, C.C.; Chaddock, J.A.; Cox, C.L.; Heaton, C.; Sutton, J.M.; Wayne, J.; Alexander, F.C.; Rogers, D.F. Re-engineering the target specificity of Clostridial neurotoxins - a route to novel therapeutics. Neurotox. Res. 2006, 9, 101–107. [Google Scholar]
- Chen, S.; Barbieri, J.T. Engineering botulinum neurotoxin to extend therapeutic intervention. Proc. Natl. Acad. Sci. USA 2009, 106, 9180–9184. [Google Scholar]
- Galli, T.; Zahraoui, A.; Vaidyanathan, V.V.; Raposo, G.; Tian, J.M.; Karin, M.; Niemann, H.; Louvard, D. A novel tetanus neurotoxin-insensitive vesicle-associated membrane protein in SNARE complexes of the apical plasma membrane of epithelial cells. Mol. Biol. Cell 1998, 9, 1437–1448. [Google Scholar]
- Chaineau, M.; Danglot, L.; Galli, T. Multiple roles of the vesicular-SNARE TI-VAMP in post-Golgi and endosomal trafficking. FEBS Lett. 2009, 583, 3817–3826. [Google Scholar]
- Sellamuthu, S.; Shin, B.H.; Lee, E.S.; Rho, S.H.; Hwang, W.; Lee, Y.J.; Han, H.E.; Kim, J.I.; Park, W.J. Engineering of protease variants exhibiting altered substrate specificity. Biochem. Biophys. Res. Commun. 2008, 371, 122–126. [Google Scholar]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [PubMed]
- Chen, S.; Hall, C.; Barbieri, J.T. Substrate recognition of VAMP-2 by botulinum neurotoxin B and tetanus neurotoxin. J. Biol. Chem. 2008, 283, 21153–21159. [Google Scholar]
- Fang, H.; Luo, W.; Henkel, J.; Barbieri, J.; Green, N. A yeast assay probes the interaction between botulinum neurotoxin serotype B and its SNARE substrate. Proc. Natl. Acad. Sci. USA 2006, 103, 6958–6963. [Google Scholar]
- Schmidt, J.J.; Bostian, K.A. Endoproteinase activity of type A botulinum neurotoxin: substrate requirements and activation by serum albumin. J. Prot. Chem. 1997, 16, 19–26. [Google Scholar]
- Shone, C.C.; Roberts, A.K. Peptide substrate specificity and properties of the zinc-endopeptidase activity of botulinum type B neurotoxin. Eur. J. Biochem. 1994, 225, 263–270. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Binz, T.; Sikorra, S.; Mahrhold, S. Clostridial Neurotoxins: Mechanism of SNARE Cleavage and Outlook on Potential Substrate Specificity Reengineering. Toxins 2010, 2, 665-682. https://doi.org/10.3390/toxins2040665
Binz T, Sikorra S, Mahrhold S. Clostridial Neurotoxins: Mechanism of SNARE Cleavage and Outlook on Potential Substrate Specificity Reengineering. Toxins. 2010; 2(4):665-682. https://doi.org/10.3390/toxins2040665
Chicago/Turabian StyleBinz, Thomas, Stefan Sikorra, and Stefan Mahrhold. 2010. "Clostridial Neurotoxins: Mechanism of SNARE Cleavage and Outlook on Potential Substrate Specificity Reengineering" Toxins 2, no. 4: 665-682. https://doi.org/10.3390/toxins2040665
APA StyleBinz, T., Sikorra, S., & Mahrhold, S. (2010). Clostridial Neurotoxins: Mechanism of SNARE Cleavage and Outlook on Potential Substrate Specificity Reengineering. Toxins, 2(4), 665-682. https://doi.org/10.3390/toxins2040665