Autoproteolytic Activation of Bacterial Toxins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Autoproteolytic Activation of Bacterial Toxins
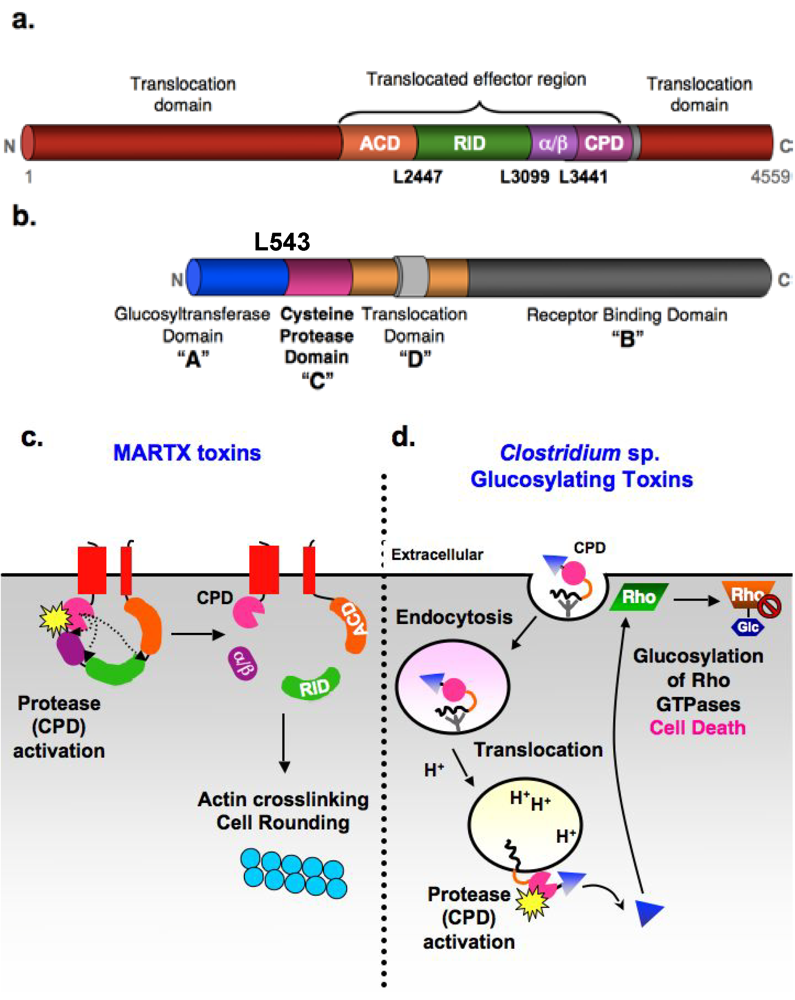
1.1. MARTX toxins

1.2. Clostridium sp. glucosylating toxins
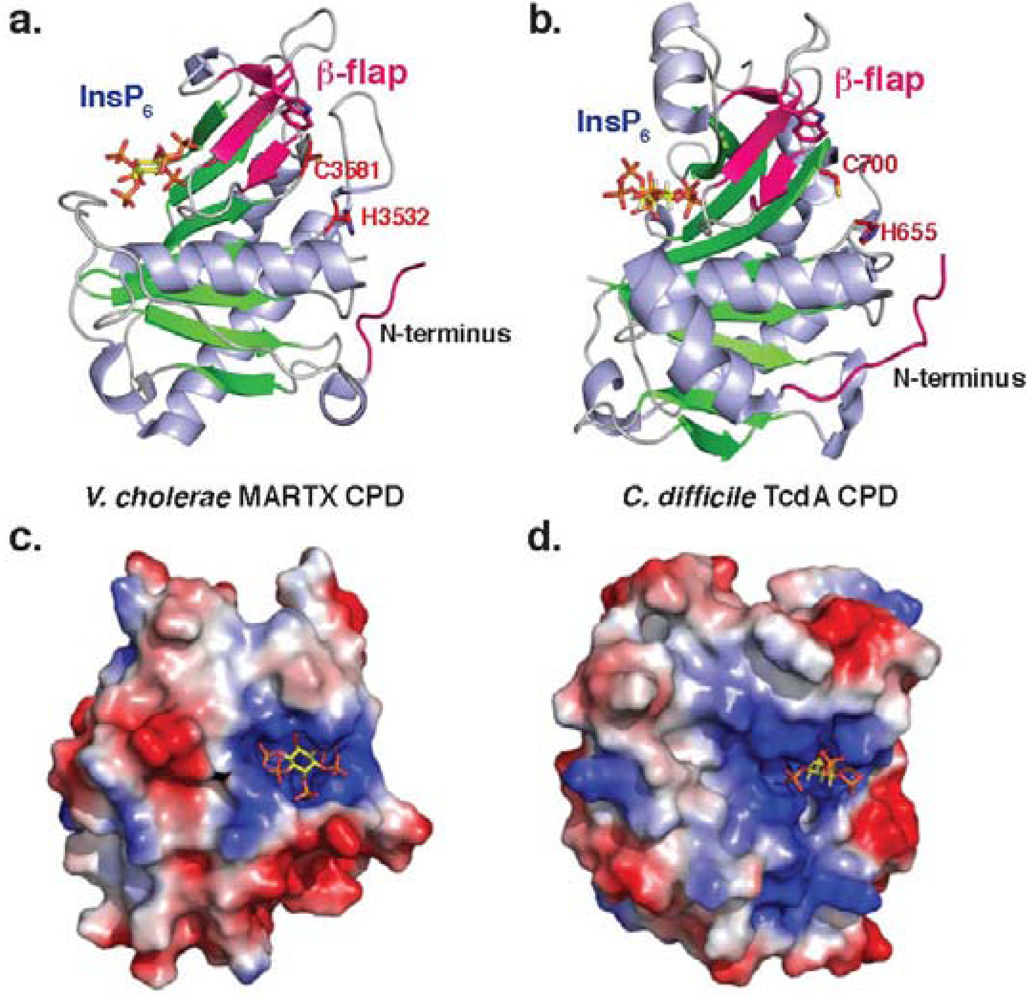
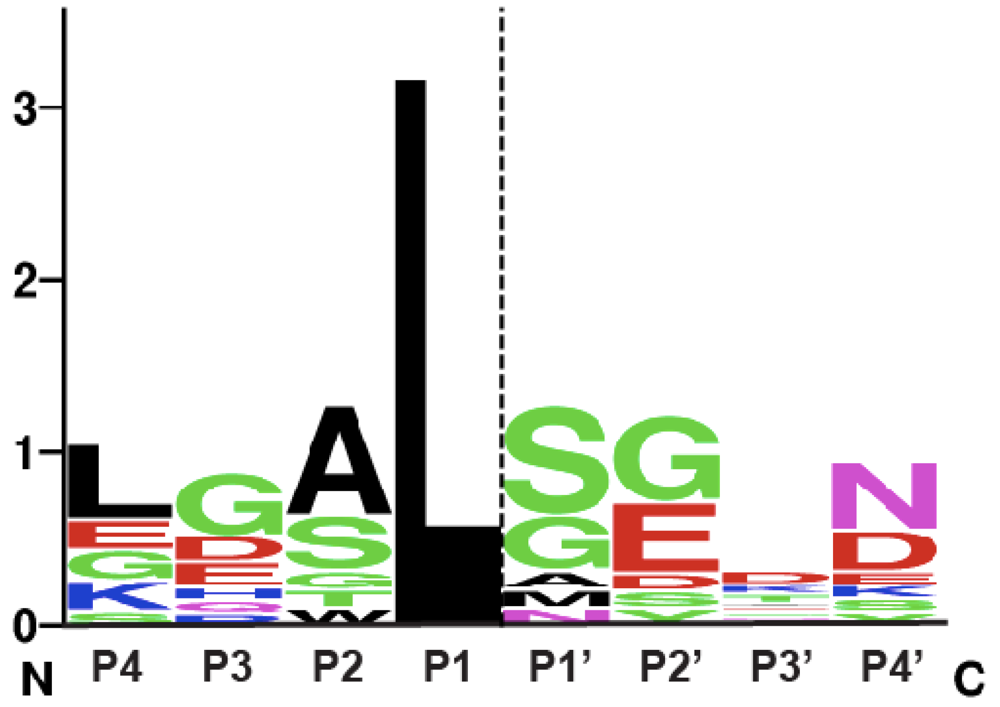
2. Autoprocessing Cysteine Protease Domains


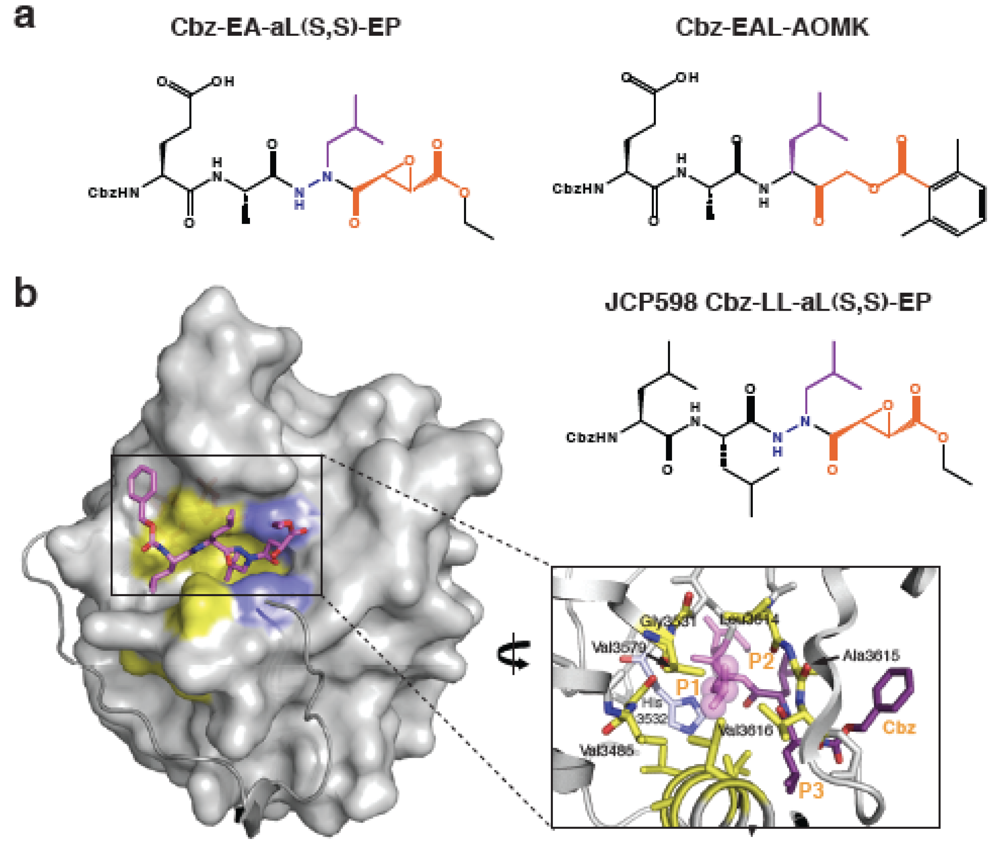
3. Development of Inhibitors of Bacterial CPD Protease Activity

4. Requirement of CPD-Mediated Processing for Toxin Activity
5. Conclusions and Perspectives
Acknowledgements
References and Notes
- Gordon, V.M.; Leppla, S.H. Proteolytic activation of bacterial toxins: role of bacterial and host cell proteases. Infect. Immun. 1994, 62, 333–340. [Google Scholar]
- Lencer, W.I.; Constable, C.; Moe, S.; Rufo, P.A.; Wolf, A.; Jobling, M.G.; Ruston, S.P.; Madara, J.L.; Holmes, R.K.; Hirst, T.R. Proteolytic activation of cholera toxin and Escherichia coli labile toxin by entry into host epithelial cells. Signal transduction by a protease-resistant toxin variant. J. Biol. Chem. 1997, 272, 15562–15568. [Google Scholar] [PubMed]
- Egerer, M.; Giesemann, T.; Jank, T.; Satchell, K.J.; Aktories, K. Auto-catalytic cleavage of Clostridium difficile toxins A and B depends on cysteine protease activity. J. Biol. Chem. 2007, 282, 25314–25321. [Google Scholar]
- Lupardus, P.J.; Shen, A.; Bogyo, M.; Garcia, K.C. Small molecule-induced allosteric activation of the vibrio cholerae rtx cysteine protease domain. Science 2008, 322, 265–268. [Google Scholar]
- Prochazkova, K.; Satchell, K.J. Structure-function analysis of inositol hexakisphosphate-induced autoprocessing of the Vibrio cholerae multifunctional autoprocessing RTX toxin. J. Biol. Chem. 2008, 283, 23656–23664. [Google Scholar] [CrossRef]
- Barroso, L.A.; Moncrief, J.S.; Lyerly, D.M.; Wilkins, T.D. Mutagenesis of the Clostridium difficile toxin B gene and effect on cytotoxic activity. Microb. Pathog. 1994, 16, 297–303. [Google Scholar]
- Jank, T.; Aktories, K. Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol. 2008, 16, 222–229. [Google Scholar] [CrossRef]
- Sheahan, K.L.; Cordero, C.L.; Satchell, K.J. Autoprocessing of the Vibrio cholerae RTX toxin by the cysteine protease domain. Embo. J. 2007, 26, 2552–2561. [Google Scholar] [CrossRef]
- Shen, A.; Lupardus, P.J.; Albrow, V.E.; Guzzetta, A.; Powers, J.C.; Garcia, K.C.; Bogyo, M. Mechanistic and structural insights into the proteolytic activation of Vibrio cholerae MARTX toxin. Nat. Chem. Biol. 2009, 5, 469–478. [Google Scholar] [CrossRef]
- Irvine, R.F.; Schell, M.J. Back in the water: the return of the inositol phosphates. Nat. Rev. Mol. Cell Biol. 2001, 2, 327–338. [Google Scholar]
- Michell, R.H. Inositol derivatives: evolution and functions. Nat. Rev. Mol. Cell. Biol. 2008, 9, 151–161. [Google Scholar] [CrossRef]
- Shears, S.B. Assessing the omnipotence of inositol hexakisphosphate. Cell Signal. 2001, 13, 151–158. [Google Scholar] [CrossRef]
- Pfeifer, G.; Schirmer, J.; Leemhuis, J.; Busch, C.; Meyer, D.K.; Aktories, K.; Barth, H. Cellular uptake of Clostridium difficile toxin B. Translocation of the N-terminal catalytic domain into the cytosol of eukaryotic cells. J. Biol. Chem. 2003, 278, 44535–44541. [Google Scholar] [PubMed]
- Satchell, K.J. MARTX, multifunctional autoprocessing repeats-in-toxin toxins. Infect. Immun. 2007, 75, 5079–5084. [Google Scholar] [CrossRef]
- Rahman, M.H.; Biswas, K.; Hossain, M.A.; Sack, R.B.; Mekalanos, J.J.; Faruque, S.M. Distribution of genes for virulence and ecological fitness among diverse Vibrio cholerae population in a cholera endemic area: tracking the evolution of pathogenic strains. DNA Cell Biol. 2008, 27, 347–355. [Google Scholar] [CrossRef]
- Chow, K.H.; Ng, T.K.; Yuen, K.Y.; Yam, W.C. Detection of RTX toxin gene in Vibrio cholerae by PCR. J. Clin. Microbiol. 2001, 39, 2594–2597. [Google Scholar]
- Cordero, C.L.; Sozhamannan, S.; Satchell, K.J. RTX toxin actin cross-linking activity in clinical and environmental isolates of Vibrio cholerae. J. Clin. Microbiol. 2007, 45, 2289–2292. [Google Scholar] [CrossRef]
- Lin, W.; Fullner, K.J.; Clayton, R.; Sexton, J.A.; Rogers, M.B.; Calia, K.E.; Calderwood, S.B.; Fraser, C.; Mekalanos, J.J. Identification of a vibrio cholerae RTX toxin gene cluster that is tightly linked to the cholera toxin prophage. Proc. Natl. Acad. Sci. USA 1999, 96, 1071–1076. [Google Scholar]
- Olivier, V.; Haines, G.K.; Satchell, K.J. Hemolysin and the multifunctional autoprocessing RTX toxin are virulence factors during intestinal infection of mice with Vibrio cholerae El Tor O1 strains. Infect. Immun. 2007, 75, 5035–5042. [Google Scholar] [CrossRef] [PubMed]
- Olivier, V.; Queen, J.; Satchell, K.J. Successful small intestine colonization of adult mice by Vibrio cholerae requires ketamine anesthesia and accessory toxins. PLoS One 2009, 4, e7352. [Google Scholar]
- Olivier, V.; Salzman, N.H.; Satchell, K.J. Prolonged colonization of mice by Vibrio cholerae El Tor O1 depends on accessory toxins. Infect. Immun. 2007, 75, 5043–5051. [Google Scholar]
- Kim, Y.R.; Lee, S.E.; Kook, H.; Yeom, J.A.; Na, H.S.; Kim, S.Y.; Chung, S.S.; Choy, H.E.; Rhee, J.H. Vibrio vulnificus RTX toxin kills host cells only after contact of the bacteria with host cells. Cell Microbiol. 2008, 10, 848–862. [Google Scholar]
- Lee, J.H.; Kim, M.W.; Kim, B.S.; Kim, S.M.; Lee, B.C.; Kim, T.S.; Choi, S.H. Identification and characterization of the Vibrio vulnificus rtxA essential for cytotoxicity in vitro and virulence in mice. J. Microbiol. 2007, 45, 146–152. [Google Scholar] [PubMed]
- Li, L.; Rock, J.L.; Nelson, D.R. Identification and characterization of a repeat-in-toxin gene cluster in Vibrio anguillarum. Infect. Immun. 2008, 76, 2620–2632. [Google Scholar]
- Prochazkova, K.; Shuvalova, L.A.; Minasov, G.; Voburka, Z.; Anderson, W.F.; Satchell, K.J. Structural and molecular mechanism for autoprocessing of MARTX toxin of Vibrio cholerae at multiple sites. J. Biol. Chem. 2009, 284, 26557–26568. [Google Scholar]
- Cordero, C.L.; Kudryashov, D.S.; Reisler, E.; Satchell, K.J. The Actin cross-linking domain of the Vibrio cholerae RTX toxin directly catalyzes the covalent cross-linking of actin. J. Biol. Chem. 2006, 281, 32366–32374. [Google Scholar]
- Geissler, B.; Bonebrake, A.; Sheahan, K.L.; Walker, M.E.; Satchell, K.J. Genetic determination of essential residues of the Vibrio cholerae actin cross-linking domain reveals functional similarity with glutamine synthetases. Mol. Microbiol. 2009, 73, 858–868. [Google Scholar]
- Kudryashov, D.S.; Cordero, C.L.; Reisler, E.; Satchell, K.J. Characterization of the enzymatic activity of the actin cross-linking domain from the Vibrio cholerae MARTX Vc toxin. J. Biol. Chem. 2008, 283, 445–452. [Google Scholar]
- Kudryashov, D.S.; Durer, Z.A.; Ytterberg, A.J.; Sawaya, M.R.; Pashkov, I.; Prochazkova, K.; Yeates, T.O.; Loo, R.R.; Loo, J.A.; Satchell, K.J.; Reisler, E. Connecting actin monomers by iso-peptide bond is a toxicity mechanism of the Vibrio cholerae MARTX toxin. Proc. Natl. Acad. Sci. USA 2008, 105, 18537–18542. [Google Scholar]
- Sheahan, K.L.; Satchell, K.J. Inactivation of small Rho GTPases by the multifunctional RTX toxin from Vibrio cholerae. Cell Microbiol. 2007, 9, 1324–1335. [Google Scholar]
- Albesa-Jove, D.; Bertrand, T.; Carpenter, E.P.; Swain, G.V.; Lim, J.; Zhang, J.; Haire, L.F.; Vasisht, N.; Braun, V.; Lange, A.; von Eichel-Streiber, C.; Svergun, D.I.; Fairweather, N.F.; Brown, K.A. Four distinct structural domains in Clostridium difficile toxin B visualized using SAXS. J. Mol. Biol. 2010, 396, 1260–1270. [Google Scholar]
- Giesemann, T.; Egerer, M.; Jank, T.; Aktories, K. Processing of Clostridium difficile toxins. J. Med. Microbiol. 2008, 57, 690–696. [Google Scholar]
- Genth, H.; Aktories, K.; Just, I. Monoglucosylation of RhoA at threonine 37 blocks cytosol-membrane cycling. J. Biol. Chem. 1999, 274, 29050–29056. [Google Scholar]
- Just, I.; Selzer, J.; Wilm, M.; von Eichel-Streiber, C.; Mann, M.; Aktories, K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 1995, 375, 500–503. [Google Scholar]
- Voth, D.E.; Ballard, J.D. Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 2005, 18, 247–263. [Google Scholar]
- Barth, H.; Pfeifer, G.; Hofmann, F.; Maier, E.; Benz, R.; Aktories, K. Low pH-induced formation of ion channels by clostridium difficile toxin B in target cells. J. Biol. Chem. 2001, 276, 10670–10676. [Google Scholar]
- Egerer, M.; Giesemann, T.; Herrmann, C.; Aktories, K. Autocatalytic processing of Clostridium difficile toxin B. Binding of inositol hexakisphosphate. J. Biol. Chem. 2009, 284, 3389–3395. [Google Scholar] [PubMed]
- Reineke, J.; Tenzer, S.; Rupnik, M.; Koschinski, A.; Hasselmayer, O.; Schrattenholz, A.; Schild, H.; von Eichel-Streiber, C. Autocatalytic cleavage of Clostridium difficile toxin B. Nature 2007, 446, 415–419. [Google Scholar]
- Kelly, C.P.; LaMont, J.T. Clostridium difficile--more difficult than ever. N. Engl. J. Med. 2008, 359, 1932–1940. [Google Scholar]
- Rupnik, M.; Wilcox, M.H.; Gerding, D.N. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 526–536. [Google Scholar] [CrossRef]
- Lyras, D.; O'Connor, J.R.; Howarth, P.M.; Sambol, S.P.; Carter, G.P.; Phumoonna, T.; Poon, R.; Adams, V.; Vedantam, G.; Johnson, S.; Gerding, D.N.; Rood, J.I. Toxin B is essential for virulence of Clostridium difficile. Nature 2009, 458, 1176–1179. [Google Scholar]
- Stabler, R.A.; Gerding, D.N.; Songer, J.G.; Drudy, D.; Brazier, J.S.; Trinh, H.T.; Witney, A.A.; Hinds, J.; Wren, B.W. Comparative phylogenomics of Clostridium difficile reveals clade specificity and microevolution of hypervirulent strains. J. Bacteriol. 2006, 188, 7297–7305. [Google Scholar]
- Rawlings, N.D.; Morton, F.R.; Kok, C.Y.; Kong, J.; Barrett, A.J. MEROPS: the peptidase database. Nucleic Acids Res. 2008, 36, D320–D325. [Google Scholar]
- Barrett, A.J.; Rawlings, N.D. Evolutionary lines of cysteine peptidases. Biol. Chem. 2001, 382, 727–733. [Google Scholar]
- Pruitt, R.N.; Chagot, B.; Cover, M.; Chazin, W.J.; Spiller, B.; Lacy, D.B. Structure-function analysis of inositol hexakisphosphate-induced autoprocessing in Clostridium difficile toxin A. J. Biol. Chem. 2009, 284, 21934–21940. [Google Scholar]
- Pop, C.; Salvesen, G.S. Human caspases: activation, specificity, and regulation. J. Biol. Chem. 2009, 284, 21777–21781. [Google Scholar] [PubMed]
- Schilling, O.; Overall, C.M. Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat. Biotechnol. 2008, 26, 685–694. [Google Scholar]
- Stennicke, H.R.; Renatus, M.; Meldal, M.; Salvesen, G.S. Internally quenched fluorescent peptide substrates disclose the subsite preferences of human caspases 1, 3, 6, 7 and 8. Biochem. J. 2000, 350 (Pt 2), 563–568. [Google Scholar] [CrossRef] [PubMed]
- Rupnik, M.; Pabst, S.; Rupnik, M.; von Eichel-Streiber, C.; Urlaub, H.; Soling, H.D. Characterization of the cleavage site and function of resulting cleavage fragments after limited proteolysis of Clostridium difficile toxin B (TcdB) by host cells. Microbiology 2005, 151, 199–208. [Google Scholar]
- Asgian, J.L.; James, K.E.; Li, Z.Z.; Carter, W.; Barrett, A.J.; Mikolajczyk, J.; Salvesen, G.S.; Powers, J.C. Aza-peptide epoxides: A new class of inhibitors selective for clan CD cysteine proteases. J. Med. Chem. 2002, 45, 4958–4960. [Google Scholar]
- Ganesan, R.; Jelakovic, S.; Campbell, A. J.; Li, Z.Z.; Asgian, J.L.; Powers, J.C.; Grutter, M.G. Exploring the S4 and S1 prime subsite specificities in caspase-3 with aza-peptide epoxide inhibitors. Biochemistry 2006, 45, 9059–9067. [Google Scholar]
- Kato, D.; Boatright, K.M.; Berger, A.B.; Nazif, T.; Blum, G.; Ryan, C.; Chehade, K.A.; Salvesen, G.S.; Bogyo, M. Activity-based probes that target diverse cysteine protease families. Nat. Chem. Biol. 2005, 1, 33–38. [Google Scholar] [CrossRef]
- Geissler, B.; Tungekar, R.; Satchell, K.J. Identification of a conserved membrane localization domain within numerous large bacterial protein toxins. Pro. Nat. Acad Sci. USA 2010, 107, 5581–5586. [Google Scholar]
- Heap, J.T.; Kuehne, S.A.; Ehsaan, M.; Cartman, S.T.; Cooksley, C.M.; Scott, J.C.; Minton, N.P. The ClosTron: Mutagenesis in Clostridium refined and streamlined. J. Microbiol. Methods 2010, 80, 49–55. [Google Scholar]
- Blair, W.S.; Semler, B.L. Self-cleaving proteases. Curr. Opin. Cell Biol. 1991, 3, 1039–1045. [Google Scholar] [CrossRef]
- Bedard, K.M.; Semler, B.L. Regulation of picornavirus gene expression. Microbes. Infect. 2004, 6, 702–713. [Google Scholar] [CrossRef]
- Lall, M.S.; Jain, R.P.; Vederas, J.C. Inhibitors of 3C cysteine proteinases from Picornaviridae. Curr. Top Med. Chem. 2004, 4, 1239–1253. [Google Scholar]
- Lee, G.M.; Craik, C.S. Trapping moving targets with small molecules. Science 2009, 324, 213–215. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shen, A. Autoproteolytic Activation of Bacterial Toxins. Toxins 2010, 2, 963-977. https://doi.org/10.3390/toxins2050963
Shen A. Autoproteolytic Activation of Bacterial Toxins. Toxins. 2010; 2(5):963-977. https://doi.org/10.3390/toxins2050963
Chicago/Turabian StyleShen, Aimee. 2010. "Autoproteolytic Activation of Bacterial Toxins" Toxins 2, no. 5: 963-977. https://doi.org/10.3390/toxins2050963
APA StyleShen, A. (2010). Autoproteolytic Activation of Bacterial Toxins. Toxins, 2(5), 963-977. https://doi.org/10.3390/toxins2050963