Toxin-Specific Antibodies for the Treatment of Clostridium difficile: Current Status and Future Perspectives †


Abstract
:1. Introduction
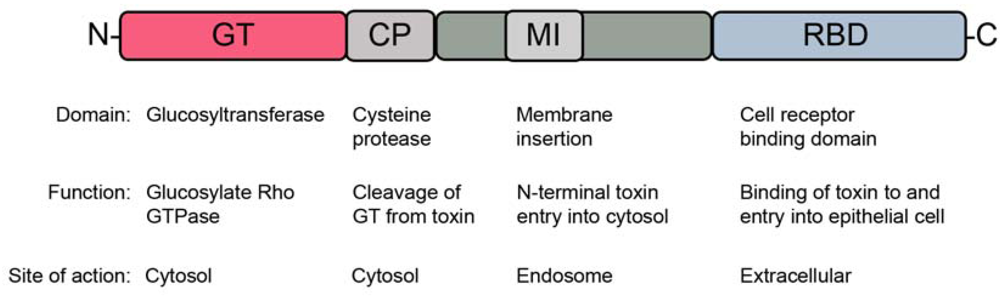
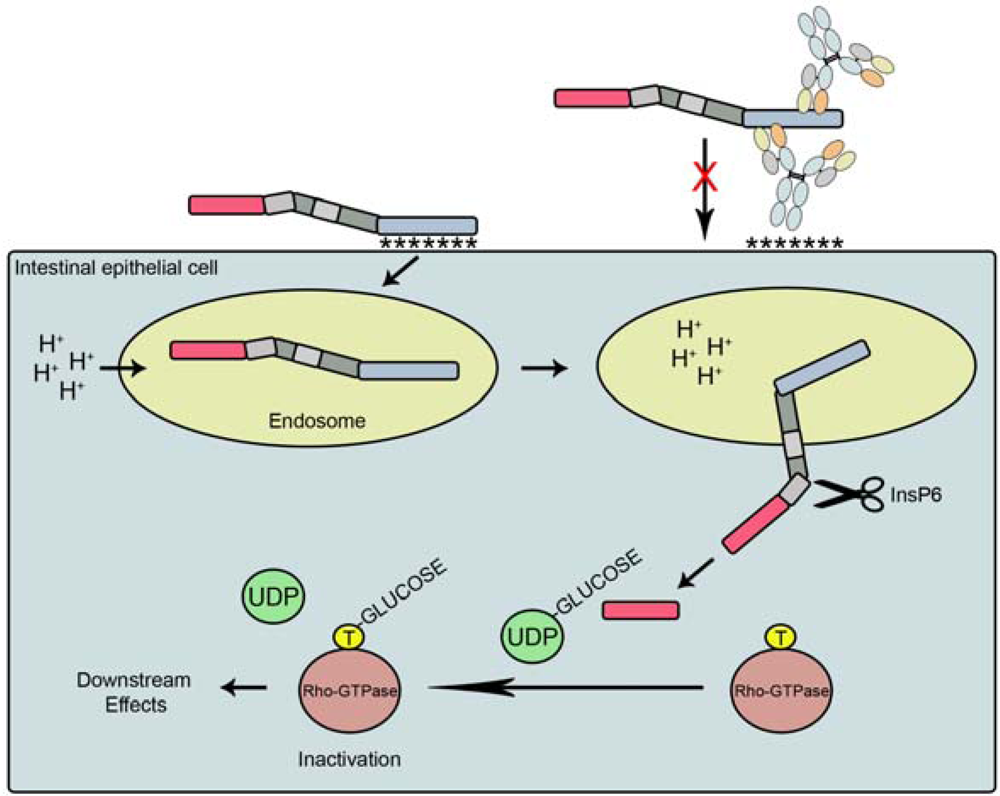
2. Toxin Structure and Function

3. Treatment of Clostridium difficile-Associated Disease


{kind=link}
{kind=link}
{kind=link}
| Type | Description | Reference |
|---|---|---|
| Antibiotic | Nitazoxanide | [34] |
| Rifaximin | [35] | |
| Ramoplanin | [36] | |
| Difimicin | [37] | |
| Probiotic | Saccharomyces boulardii | [38] |
| Lactobacillus spp. | [39] | |
| Fecal transplantation | Stool replacement therapy | [40] |
| Toxin binding agents | Cholestyramine | [41] |
| Tolevamer | [42] | |
| Vaccine | Toxoid-based | [43] |
| SLP-based | [44] | |
| DNA-based | [45] | |
| Antibodies | IgG, IgA, IgY, polyclonal | See Table 2 and Table 3 |
| scFv | [46] | |
| sdAb | [47] |
4. Toxin-Specific Antibodies
4.1. Role of antibodies in CDAD
4.2. Experimental animal studies

| Antibody | Specificity | Immunogen | Antibody Source | Animal Model | Challenge Type | Ab Administration Route | Treatment Type | Outcome | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| PCG-4 IgG | TcdA | Culture filtrate | Mouse | Hamster | Oral TcdA administration | Oral | Ab + TcdA co-administered | Protection | [60] |
| G-2 IgG | TcdA and TcdB | Toxoid B | Mouse hybridoma | Hamster | Oral TcdA administration | Oral | Ab + TcdA co-administered | No protection | [60] |
| 37B5 IgG | TcdA | Toxoid A | Hybridoma | Mouse | I.P. TcdA administration | I.P. | Ab + TcdA co-administered | No protection | [61] |
| A9, 141-2, C11 IgGs | TcdA | Toxoid A | Mouse | Mouse | Oral C. difficile1 | I.V. | Prophylactic | Protection | [62] |
| Bovine Ig | TcdA and TcdB | Culture filtrate | Cow colostrum | Hamster | Oral C. difficile (108 cells) | Oral | Prophylactic | Protection | [63] |
| Bovine Ig | TcdA and TcdB | Culture filtrate | Cow colostrum | Rat | CD filtrate into ileum 2 | Ileum injection 2 | Ab + toxin co-injected 2 | Protection | [64] |
| Anti-TcdA Bovine Ig | TcdA | Toxoid A | Cow colostrum | Rat | CD filtrate into ileum 2 | Ileum injection 2 | Ab + toxin co-injected 2 | Protection | [64] |
| Anti-TcdA IgY | TcdA | rTcdA fragment | Chicken | Hamster | Oral C. difficile (104 cells) | Oral | Treatment and relapse | Protection | [65] |
| Anti-TcdB IgY | TcdB | rTcdB fragment | Chicken | Hamster | Oral C. difficile (104 cells) | Oral | Treatment and relapse | Protection | [65] |
| Polyclonal | TcdA and TcdB | rTcdA/B toxoid | Mouse | Hamster | Oral C. difficile (105 cells) | I.P. | Prophylactic | Protection | [66] |
| Bovine immune whey | TcdA and TcdB | Culture filtrate | Cow | Hamster | Oral C. difficile (104 cells) | Oral | Prophylactic and treatment | Protection | [67] |
| CDA1 IgG | TcdA | Toxoid A | Mouse hybridoma 3 | Hamster | Oral C. difficile spores (140) 4 | I.P. | Treatment and relapse | Protection | [7] |
| MDX-1388 IgG | TcdB | rTcdB fragment | Mouse hybridoma 3 | Hamster | Oral C. difficile spores (140K) 4 | I.P. | Treatment and relapse | Protection | [7] |
| Antibody | Specificity | Source | Administration Route | Number of Treated Patients | Treatment Success Rate (%) | Ref |
|---|---|---|---|---|---|---|
| IVIG prep | TcdA | Human | I.V. | 5 | 100 | [53] |
| IVIG prep | TcdA and TcdB | Human | I.V. | 2 | 100 | [68] |
| IVIG prep | unknown | Human | I.V. | 4 | 100 | [69] |
| IVIG prep | unknown | Human | I.V. | 5 | 60 | [70] |
| IVIG prep | unknown | Human | I.V. | 14 | 64 | [71] |
| IVIG prep | unknown | Human | I.V. | 1 | 100 | [72] |
| IVIG prep | unknown | Human | I.V. | 18 | 83 | [76] |
| IVIG prep | unknown | Human | I.V. | 1 | 100 | [73] |
| IVIG prep | unknown | Human | I.V. | 1 | 100 | [74] |
| IVIG prep | unknown | Human | I.V. | 2 | 100 | [75] |
| IVIG prep | unknown | Human | I.V. | 21 | 43 | [77] |
| IgA | unknown | Human | Oral | 1 | 100 | [78] |
| Bovine immune whey | TcdA and TcdB | Cow | Oral | 15 | 93 | [67] |
| Bovine immune whey | TcdA and TcdB | Cow | Oral | 101 | 90 | [79] |
| Bovine immune whey | TcdA and TcdB | Cow | Oral | 20 | 55 | [80] |
| CDA1 IgG | TcdA | Mouse hybridoma 1 | I.V. | 101 | 93 | [81] |
| CDB1 IgG | TcdB | Mouse hybridoma 1 | I.V. | 101 | 93 | [81] |
4.3. Experimental human studies
4.4. Antibody mechanism of action
5. Future Perspectives for CDAD Immunotherapy
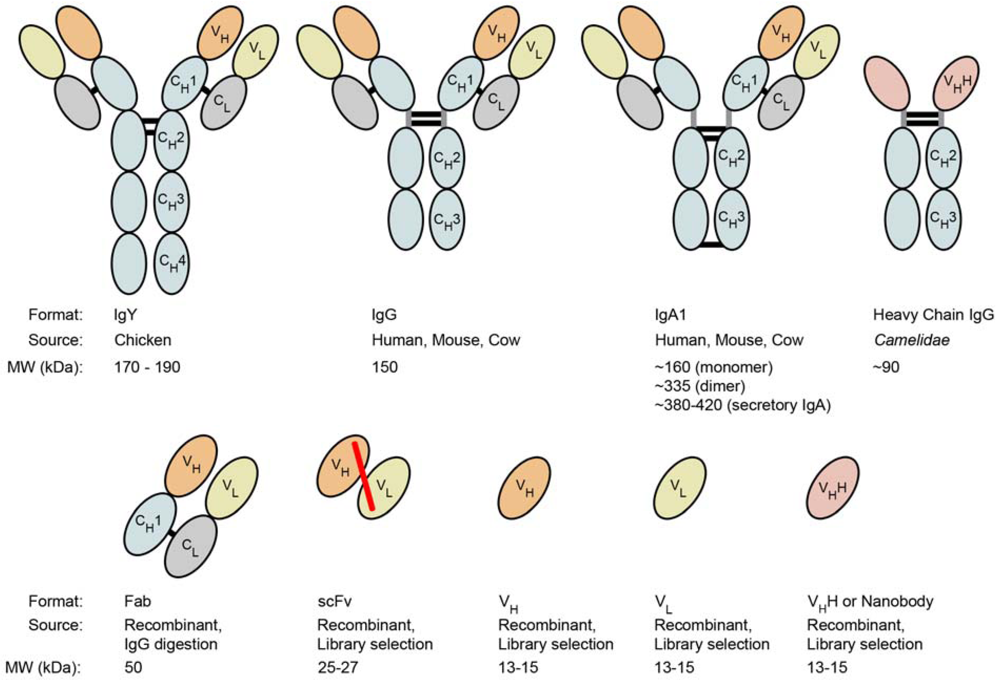
5.1. Recombinant antibody fragments
5.2. Single-domain antibodies
6. Conclusions
References
- Rupnik, M.; Wilcox, M.H.; Gerding, D.N. Clostridium difficile infection: New developments in epidemiology and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 526–536. [Google Scholar]
- Leffler, D.A.; Lamont, J.T. Treatment of Clostridium difficile-associated disease. Gastroenterology 2009, 136, 1899–1912. [Google Scholar] [CrossRef]
- O’Connor, J.R.; Johnson, S.; Gerding, D.N. Clostridium difficile infection caused by the epidemic BI/NAP1/027 strain. Gastroenterology 2009, 136, 1913–1924. [Google Scholar]
- McDonald, L.C.; Killgore, G.E.; Thompson, A.; Owens, R.C. Jr.; Kazakova, S.V.; Sambol, S.P.; Johnson, S.; Gerding, D.N. An epidemic, toxin gene-variant strain of Clostridium difficile. N. Engl. J. Med. 2005, 353, 2433–2441. [Google Scholar]
- Warny, M.; Pepin, J.; Fang, A.; Killgore, G.; Thompson, A.; Brazier, J.; Frost, E.; McDonald, L.C. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet 2005, 366, 1079–1084. [Google Scholar]
- Lyras, D.; O’Connor, J.R.; Howarth, P.M.; Sambol, S.P.; Carter, G.P.; Phumoonna, T.; Poon, R.; Adams, V.; Vedantam, G.; Johnson, S.; Gerding, D.N.; Rood, J.I. Toxin B is essential for virulence of Clostridium difficile. Nature 2009, 458, 1176–1179. [Google Scholar]
- Babcock, G.J.; Broering, T.J.; Hernandez, H.J.; Mandell, R.B.; Donahue, K.; Boatright, N.; Stack, A.M.; Lowy, I.; Graziano, R.; Molrine, D.; Ambrosino, D.M.; Thomas, W.D. Jr. Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect. Immun. 2006, 74, 6339–6347. [Google Scholar]
- Kim, P.H.; Iaconis, J.P.; Rolfe, R.D. Immunization of adult hamsters against Clostridium difficile-associated ileocecitis and transfer of protection to infant hamsters. Infect. Immun. 1987, 55, 2984–2992. [Google Scholar]
- Lyerly, D.M.; Saum, K.E.; MacDonald, D.K.; Wilkins, T.D. Effects of Clostridium difficile toxins given intragastrically to animals. Infect. Immun. 1985, 47, 349–352. [Google Scholar]
- Rasko, D.A.; Sperandio, V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar]
- Bebbington, C.; Yarranton, G. Antibodies for the treatment of bacterial infections: current experience and future prospects. Curr. Opin. Biotechnol. 2008, 19, 613–619. [Google Scholar] [CrossRef]
- Clatworthy, A.E.; Pierson, E.; Hung, D.T. Targeting virulence: a new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. [Google Scholar] [CrossRef]
- Jank, T.; Aktories, K. Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol. 2008, 16, 222–229. [Google Scholar] [CrossRef]
- Voth, D.E.; Ballard, J.D. Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 2005, 18, 247–263. [Google Scholar]
- Jank, T.; Giesemann, T.; Aktories, K. Rho-glucosylating Clostridium difficile toxins A and B: new structural insights into structure and function. Glycobiology 2007, 17, 15R–22R. [Google Scholar] [CrossRef]
- Pothoulakis, C. Effects of Clostridium difficile toxins on epithelial cell barrier. Ann. N.Y. Acad. Sci. 2000, 915, 347–356. [Google Scholar] [CrossRef]
- Calabi, E.; Calabi, F.; Phillips, A.D.; Fairweather, N.F. Binding of Clostridium difficile surface layer proteins to gastrointestinal tissues. Infect. Immun. 2002, 70, 5770–5778. [Google Scholar]
- Lee, A.S.; Song, K.P. LuxS/autoinducer-2 quorum sensing molecule regulates transcriptional virulence gene expression in Clostridium difficile. Biochem. Biophys. Res. Commun. 2005, 335, 659–666. [Google Scholar] [CrossRef]
- Albesa-Jové, D.; Bertrand, T.; Carpenter, E.P.; Swain, G.V.; Lim, J.; Zhang, J.; Haire, L.F.; Vasisht, N.; Braun, V.; Lange, A.; von Eichel-Streiber, C.; Svergun, D.I.; Fairweather, N.F.; Brown, K.A. Four distinct structural domains in Clostridium difficile toxin B visualized using SAXS. J. Mol. Biol. 2010, 396, 1260–1270. [Google Scholar]
- Dingle, T.; Wee, S.; Mulvey, G.L.; Greco, A.; Kitova, E.N.; Sun, J.; Lin, S.; Klassen, J.S.; Palcic, M.M.; Ng, K.K.; Armstrong, G.D. Functional properties of the carboxy-terminal host cell-binding domains of the two toxins, TcdA and TcdB, expressed by Clostridium difficile. Glycobiology 2008, 18, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; Ho, J.G.; Lin, S.J.; Palcic, M.M.; Rupnik, M.; Ng, K.K. Carbohydrate recognition by Clostridium difficile toxin A. Nat. Struct. Mol. Biol. 2006, 13, 460–461. [Google Scholar] [CrossRef]
- Ho, J.G.; Greco, A.; Rupnik, M.; Ng, K.K. Crystal structure of receptor-binding C-terminal repeats from Clostridium difficile toxin A. Proc. Natl. Acad. Sci. USA 2005, 102, 18373–18378. [Google Scholar]
- Krivan, H.C.; Clark, G.F.; Smith, D.F.; Wilkins, T.D. Cell surface binding site for Clostridium difficile enterotoxin: evidence for a glycoconjugate containing the sequence Gal alpha 1-3Gal beta 1-4GlcNAc. Infect. Immun. 1986, 53, 573–581. [Google Scholar]
- Reineke, J.; Tenzer, S.; Rupnik, M.; Koschinski, A.; Hasselmayer, O.; Schrattenholz, A.; Schild, H.; von Eichel-Streiber, C. Autocatalytic cleavage of Clostridium difficile toxin B. Nature 2007, 446, 415–419. [Google Scholar]
- Egerer, M.; Giesemann, T.; Jank, T.; Satchell, K.J.; Aktories, K. Auto-catalytic cleavage of Clostridium difficile toxins A and B depends on cysteine protease activity. J. Biol. Chem. 2007, 282, 25314–25321. [Google Scholar]
- Just, I.; Selzer, J.; Wilm, M,; von Eichel-Streiber, C.; Mann, M.; Aktories, K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 1995, 375, 500–503. [Google Scholar] [PubMed]
- Hecht, G.; Koutsouris, A.; Pothoulakis, C.; LaMont, J.T.; Madara, J.L. Clostridium difficile toxin B disrupts the barrier function of T84 monolayers. Gastroenterology 1992, 102, 416–423. [Google Scholar]
- Peláez, T.; Alcalá, L.; Alonso, R.; Rodríguez-Créixems, M.; García-Lechuz, J.M.; Bouza, E. Reassessment of Clostridium difficile susceptibility to metronidazole and vancomycin. Antimicrob. Agents Chemother. 2002, 46, 1647–1650. [Google Scholar]
- Gerding, D.N. Metronidazole for Clostridium difficile-associated disease: is it okay for Mom? Clin. Infect. Dis. 2005, 40, 1598–1600. [Google Scholar] [CrossRef]
- Bauer, M.P.; van Dissel, J.T.; Kuijper, E.J. Clostridium difficile: controversies and approaches to management. Curr. Opin. Infect. Dis. 2009, 22, 517–524. [Google Scholar]
- Johnson, S. Recurrent Clostridium difficile infection: a review of risk factors, treatments, and outcome. J. Infect. 2009, 58, 403–410. [Google Scholar] [CrossRef]
- Parkes, G.C.; Sanderson, J.D.; Whelan, K. The mechanisms and efficacy of probiotics in the prevention of Clostridium difficile-associated diarrhoea. Lancet Infect. Dis. 2009, 9, 237–244. [Google Scholar] [CrossRef]
- O’Horo, J.; Safdar, N. The role of immunoglobulin for the treatment of Clostridium difficile infection: a systemic review. Int. J. Infect. Dis. 2009, 13, 663–667. [Google Scholar]
- Musher, D.M.; Logan, N.; Hamill, R.J.; Dupont, H.L.; Lentnek, A.; Gupta, A.; Rossignol, J.F. Nitazoxanide for the treatment of Clostridium difficile colitis. Clin. Infect. Dis. 2006, 43, 421–427. [Google Scholar]
- Garey, K.W.; Salazar, M.; Shah, D.; Rodrigue, R.; DuPont, H.L. Rifamycin antibiotics for treatment of Clostridium difficile-associated diarrhea. Ann. Pharmacother. 2008, 42, 827–835. [Google Scholar] [CrossRef]
- Farver, D.K.; Hedge, D.D.; Lee, S.C. Ramoplanin: a lipoglycodepsipeptide antibiotic. Ann. Pharmacother. 2005, 39, 863–868. [Google Scholar]
- Sullivan, K.M.; Spooner, L.M. Fidaxomicin: a macrocyclic antibiotic for the management of Clostridium difficile infection. Ann. Pharmacother. 2010, 44, 352–359. [Google Scholar] [CrossRef]
- Tung, J.M.; Dolovich, L.R.; Lee, C.H. Prevention of Clostridium difficile infection with Saccharomyces boulardii: a systemic review. Can. J. Gastroenterol. 2009, 23, 817–821. [Google Scholar]
- Guarino, A.; Lo Vecchio, A.; Canani, R.B. Probiotics as prevention and treatment for diarrhea. Curr. Opin. Gastroenterol. 2009, 25, 18–23. [Google Scholar]
- Aas, J.; Gessert, C.E.; Bakken, J.S. Recurrent Clostridium difficile colitis: case series involving 18 patients treated with donor stool administered via a nasogastric tube. Clin. Infect. Dis. 2003, 36, 580–585. [Google Scholar] [CrossRef]
- Moncino, M.D.; Falletta, J.M. Multiple relapses of Clostridium difficile-associated diarrhea in a cancer patient. Successful control with long-term cholestyramine therapy. Am. J. Pediatr. Hematol. Oncol. 1992, 14, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Scheinfeld, N.; Biggers, K. Tolevamer, an orally administered, toxin-binding polymer for Clostridium difficile-associated diarrhea. Curr. Opin. Investig. Drugs 2008, 9, 913–924. [Google Scholar] [PubMed]
- Sougioultzis, S.; Kyne, L.; Drudy, D.; Keates, S.; Maroo, S.; Pothoulakis, C.; Giannasca, P.J.; Lee, C.K.; Warny, M.; Monath, T.P.; Kelly, C.P. Clostridium difficile toxoid vaccine in recurrent C. difficile-associated diarrhea. Gastroenterology 2005, 128, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Ní Eidhin, D.B.; O’Brien, J.B.; McCabe, M.S.; Athié-Morales, V.; Kelleher, D.P. Active immunization of hamsters against Clostridium difficile infection using surface-layer protein. FEMS Immunol. Med. Microbiol. 2008, 52, 207–218. [Google Scholar] [CrossRef]
- Gardiner, D.F.; Rosenberg, T.; Zaharatos, J.; Franco, D.; Ho, D.D. A DNA vaccine targeting the receptor-binding domain of Clostridium difficile toxin A. Vaccine 2009, 27, 3598–3604. [Google Scholar]
- Deng, X.K.; Nesbit, L.A.; Morrow, K.J. Jr. Recombinant single-chain variable fragment antibodies directed against Clostridium difficile toxin B produced by use of an optimized phage display system. Clin. Diagn. Lab Immunol. 2003, 10, 587–595. [Google Scholar]
- Hussack, G.; Arbabi-Ghahroudi, M.; van Faassen, H.; Songer, J.G.; MacKenzie, R.; Tanha, J. Manuscript in preparation. 2010. [Google Scholar]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Kyne, L.; Warny, M.; Qamar, A.; Kelly, C.P. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet 2001, 357, 189–193. [Google Scholar]
- Warny, M.; Vaerman, J.P.; Avesani, V.; Delmée, M. Human antibody response to Clostridium difficile toxin A in relation to clinical course of infection. Infect. Immun. 1994, 62, 384–389. [Google Scholar]
- Viscidi, R.; Laughon, B.E.; Yolken, R.; Bo-Linn, P.; Moench, T.; Ryder, R.W.; Bartlett, J.G. Serum antibody response to toxins A and B of Clostridium difficile. J. Infect. Dis. 1983, 148, 93–100. [Google Scholar] [CrossRef]
- Katchar, K.; Taylor, C.P.; Tummala, S.; Chen, X.; Sheikh, J.; Kelly, C.P. Association between IgG2 and IgG3 subclass responses to toxin A and recurrent Clostridium difficile-associated disease. Clin. Gastroenterol. Hepatol. 2007, 5, 707–713. [Google Scholar]
- Leung, D.Y.; Kelly, C.P.; Boguniewicz, M.; Pothoulakis, C.; LaMont, J.T.; Flores, A. Treatment with intravenously administered gamma globulin of chronic relapsing colitis induced by Clostridium difficile toxin. J. Pediatr. 1991, 118, 633–637. [Google Scholar] [CrossRef]
- Johal, S.S.; Lambert, C.P.; Hammond, J.; James, P.D.; Borriello, S.P.; Mahida, Y.R. Colonic IgA producing cells and macrophages are reduced in recurrent and non-recurrent Clostridium difficile associated diarrhoea. J. Clin. Pathol. 2004, 57, 973–979. [Google Scholar]
- Kelly, C.P.; Pothoulakis, C.; Orellana, J.; LaMont, J.T. Human colonic aspirates containing immunoglobulin A antibody to Clostridium difficile toxin A inhibit toxin A-receptor binding. Gastroenterology 1992, 102, 35–40. [Google Scholar]
- Johnson, S.; Sypura, W.D.; Gerding, D.N.; Ewing, S.L.; Janoff, E.N. Selective neutralization of a bacterial enterotoxin by serum immunoglobulin A in response to mucosal disease. Infect. Immun. 1995, 63, 3166–3173. [Google Scholar]
- Giannasca, P.J.; Warny, M. Active and passive immunization against Clostridium difficile diarrhea and colitis. Vaccine 2004, 22, 848–856. [Google Scholar] [CrossRef]
- Kim, P.H.; Rolfe, R.D. Characterization of protective antibodies in hamsters immunized against C. difficile toxins A and B. Microb. Ecol. Health Dis. 1989, 2, 47–59. [Google Scholar] [CrossRef]
- Allo, M.; Silva, J., Jr.; Fekety, R.; Rifkin, G.D.; Waskin, H. Prevention of clindamycin-induced colitis in hamsters by Clostridium sordellii antitoxin. Gastroenterology 1979, 76, 351–355. [Google Scholar]
- Lyerly, D.M.; Phelps, C.J.; Toth, J.; Wilkins, T.D. Characterization of toxins A and B of Clostridium difficile with monoclonal antibodies. Infect. Immun. 1986, 54, 70–76. [Google Scholar]
- Kamiya, S.; Yamakawa, K.; Meng, X.Q.; Ogura, H.; Nakamura, S. Production of monoclonal antibody to Clostridium sordellii toxin A which neutralizes enterotoxicity but not haemagglutination activity. FEMS Microbiol. Lett. 1991, 81, 311–316. [Google Scholar]
- Corthier, G.; Muller, M.C.; Wilkins, T.D.; Lyerly, D.; L’Haridon, R. Protection against experimental pseudomembranous colitis in gnotobiotic mice by use of monoclonal antibodies against Clostridium difficile toxin A. Infect. Immun. 1991, 59, 1192–1195. [Google Scholar]
- Lyerly, D.M.; Bostwick, E.F.; Binion, S.B.; Wilkins, T.D. Passive immunization of hamsters against disease caused by Clostridium difficile by use of bovine immunoglobulin G concentrate. Infect. Immun. 1991, 59, 2215–2218. [Google Scholar]
- Kelly, C.P.; Pothoulakis, C.; Vavva, F.; Castagliuolo, I.; Bostwick, E.F.; O’Keane, J.C.; Keates, S.; LaMont, J.T. Anti-Clostridium difficile bovine immunoglobulin concentrate inhibits cytotoxicity and enterotoxicity of C. difficile toxins. Antimicrob. Agents Chemother. 1996, 40, 373–379. [Google Scholar] [PubMed]
- Kink, J.A.; Williams, J.A. Antibodies to recombinant Clostridium difficile toxins A and B are an effective treatment and prevent relapse of C. difficile-associated disease in a hamster model of infection. Infect. Immun. 1998, 66, 2018–2025. [Google Scholar] [PubMed]
- Giannasca, P.J.; Zhang, Z.X.; Lei, W.D.; Boden, J.A.; Giel, M.A.; Monath, T.P.; Thomas, W.D. Jr. Serum antitoxin antibodies mediate systemic and mucosal protection from Clostridium difficile disease in hamsters. Infect. Immun. 1999, 67, 527–538. [Google Scholar]
- van Dissel, J.T.; de Groot, N.; Hensgens, C.M.; Numan, S.; Kuijper, E.J.; Veldkamp, P.; van ’t Wout, J. Bovine antibody-enriched whey to aid in the prevention of a relapse of Clostridium difficile-associated diarrhoea: preclinical and preliminary clinical data. J. Med. Microbiol. 2005, 54, 197–205. [Google Scholar] [CrossRef]
- Salcedo, J.; Keates, S.; Pothoulakis, C.; Warny, M.; Castagliuolo, I.; LaMont, J.T.; Kelly, C.P. Intravenous immunoglobulin therapy for severe Clostridium difficile colitis. Gut 1997, 41, 366–370. [Google Scholar]
- Beales, I.L. Intravenous immunoglobulin for recurrent Clostridium difficile diarrheoea. Gut 2002, 51, 456. [Google Scholar] [CrossRef]
- Wilcox, M.H. Descriptive study of intravenous immunoglobulin for the treatment of recurrent Clostridium difficile diarrhoea. J. Antimicrob. Chemother. 2004, 53, 882–884. [Google Scholar] [CrossRef]
- McPherson, S.; Rees, C.J.; Ellis, R.; Soo, S.; Panter, S.J. Intravenous immunoglobulin for the treatment of severe, refractory, and recurrent Clostridium difficile diarrhea. Dis. Colon Rectum 2006, 49, 640–645. [Google Scholar] [PubMed]
- Murphy, C.; Vernon, M.; Cullen, M. Intravenous immunoglobulin for resistant Clostridium difficile infection. Age Ageing 2006, 35, 85–86. [Google Scholar] [CrossRef]
- Hassoun, A.; Ibrahim, F. Use of intravenous immunoglobulin for the treatment of severe Clostridium difficile colitis. Am. J. Geriatr. Pharmacother. 2007, 5, 48–51. [Google Scholar] [CrossRef]
- Koulaouzidis, A.; Tatham, R.; Moschos, J.; Tan, C.W. Successful treatment of Clostridium difficile colitis with intravenous immunoglobulin. J. Gastrointestin. Liver Dis. 2008, 17, 353–355. [Google Scholar]
- Chandrasekar, T.; Naqvi, N.; Waddington, A.; Cooke, R.P.D.; Anijeet, H.; Gradden, C.W.; Abraham, K.A.; Wong, C.F. Intravenous immunoglobulin therapy for refractory Clostridium difficile toxin colitis in chronic kidney disease: case reports and literature review. NDT Plus 2008, 1, 20–22. [Google Scholar]
- Juang, P.; Skledar, S.J.; Zgheib, N.K.; Paterson, D.L.; Vergis, E.N.; Shannon, W.D.; Ansani, N.T.; Branch, R.A. Clinical outcomes of intravenous immune globulin in severe Clostridium difficile-associated diarrhea. Am. J. Infect. Control 2007, 35, 131–137. [Google Scholar]
- Abougergi, M.S.; Broor, A.; Cui, W.; Jaar, B.G. Intravenous immunoglobulin for the treatment of severe Clostridium difficile colitis: an observational study and review of the literature. J. Hospital Med. 2010, 5, E1–E9. [Google Scholar]
- Tjellström, B.; Stenhammar, L.; Eriksson, S.; Magnusson, K.E. Oral immunoglobulin A supplement in treatment of Clostridium difficile enteritis. Lancet 1993, 341, 701–702. [Google Scholar]
- Numan, S.C.; Veldkamp, P.; Kuijper, E.J.; van den Berg, R.J.; van Dissel, J.T. Clostridium difficile-associated diarrhoea: bovine anti-Clostridium difficile whey protein to help aid the prevention of relapses. Gut 2007, 56, 888–889. [Google Scholar]
- Mattila, E.; Anttila, V.J.; Broas, M.; Marttila, H.; Poukka, P.; Kuusisto, K.; Pusa, L.; Sammalkorpi, K.; Dabek, J.; Koivurova, O.P.; Vähätalo, M.; Moilanen, V.; Widenius, T. A randomized, double-blind study comparing Clostridium difficile immune whey and metronidazole for recurrent Clostridium difficile-associated diarrhoea: efficacy and safety data of a prematurely interrupted trial. Scand. J. Infect. Dis. 2008, 40, 702–708. [Google Scholar]
- Lowy, I.; Molrine, D.C.; Leav, B.A.; Blair, B.M.; Baxter, R.; Gerding, D.N.; Nichol, G.; Thomas, W.D. Jr.; Leney, M.; Sloan, S.; Hay, C.A.; Ambrosino, D.M. Treatment with monoclonal antibodies against Clostridium difficile toxins. N. Engl. J. Med. 2010, 362, 197–205. [Google Scholar]
- Kyne, L. Clostridium difficile-beyond antibiotics. N. Engl. J. Med. 2010, 362, 264–265. [Google Scholar] [CrossRef]
- Yoshida, M.; Kobayashi, K.; Kuo, T.T.; Bry, L.; Glickman, J.N.; Claypool, S.M.; Kaser, A.; Nagaishi, T.; Higgins, D.E.; Mizoguchi, E.; Wakatsuki, Y.; Roopenian, D.C.; Mizoguchi, A.; Lencer, W.I.; Blumberg, R.S. Neonatal Fc receptor for IgG regulates mucosal immune responses to luminal bacteria. J. Clin. Invest. 2006, 116, 2142–2151. [Google Scholar]
- Warny, M.; Fatimi, A.; Bostwick, E.F.; Laine, D.C.; Lebel, F.; LaMont, J.T.; Pothoulakis, C.; Kelly, C.P. Bovine immunoglobulin concentrate-Clostridium difficile retains C. difficile toxin neutralising activity after passage through the human stomach and small intestine. Gut 1999, 44, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboom, H.R. Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 2005, 23, 1105–1116. [Google Scholar] [CrossRef]
- Powers, D.B.; Amersdorfer, P.; Poul, M.; Nielsen, U.B.; Shalaby, M.R.; Adams, G.P.; Marks, J.D. Expression of single-chain Fv-Fc fusions in Pichia pastoris. J. Immunol. Methods 2001, 251, 123–135. [Google Scholar] [CrossRef]
- Zhang, J.; MacKenzie, R.; Durocher, Y. Production of chimeric heavy-chain antibodies. Methods Mol. Biol. 2009, 525, 323–336. [Google Scholar] [CrossRef]
- Van Bockstaele, F.; Holz, J.B.; Revets, H. The development of nanobodies for therapeutic applications. Curr. Opin. Investig. Drugs 2009, 10, 1212–1224. [Google Scholar]
- Wesolowski, J.; Alzogaray, V.; Reyelt, J.; Unger, M.; Juarez, K.; Urrutia, M.; Cauerhff, A.; Danguah, W.; Rissiek, B.; Scheuplein, F.; Schwarz, N.; Adriouch, S.; Boyer, O.; Seman, M.; Licea, A.; Serreze, D.V.; Goldbaum, F.A.; Haag, F.; Koch-Nolte, F. Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med. Microbiol. Immunol. 2009, 198, 157–174. [Google Scholar]
- Ryan, S.; Kell, A.J.; van Faassen, H.; Tay, L.L.; Simard, B.; MacKenzie, R.; Gilbert, M.; Tanha, J. Single-domain antibody-nanoparticles: promising architectures for increased Staphylococcus aureus detection specificity and sensitivity. Bioconjug. Chem. 2009, 20, 1966–1974. [Google Scholar]
- To, R.; Hirama, T.; Arbabi-Ghahroudi, M.; MacKenzie, R.; Wang, P.; Xu, P.; Ni, F.; Tanha, J. Isolation of monomeric human V(H)s by a phage selection. J. Biol. Chem. 2005, 280, 41395–41403. [Google Scholar]
- Holt, L.J.; Herring, C.; Jespers, L.S.; Woolven, B.P.; Tomlinson, I.M. Domain antibodies: proteins for therapy. Trends Biotechnol. 2003, 21, 484–490. [Google Scholar] [CrossRef]
- Ward, E.S.; Güssow, D.; Griffiths, A.D.; Jones, P.T.; Winter, G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature 1989, 341, 544–546. [Google Scholar]
- De Genst, E.; Saerens, D.; Muyldermans, S.; Conrath, K. Antibody repertoire development in camelids. Dev. Comp. Immunol. 2006, 30, 187–198. [Google Scholar] [CrossRef]
- Arbabi-Ghahroudi, M.; Desmyter, A.; Wyns, L.; Hamers, R.; Muyldermans, S. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett. 1997, 414, 521–526. [Google Scholar]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar]
- Dooley, H.; Flajnik, M.F. Antibody repertoire development in cartilaginous fish. Dev. Comp. Immunol. 2006, 30, 43–56. [Google Scholar] [CrossRef]
- Harmsen, M.M.; De Haard, H.J. Properties, production, and applications of camelid single-domain antibody fragment. Appl. Microbiol. Biotechnol. 2007, 77, 13–22. [Google Scholar] [CrossRef]
- Harmsen, M.M.; van Solt, C.B.; van Zijderveld-van Bemmel, A.M.; Niewold, T.A.; van Zijderveld, F.G. Selection and optimization of proteolytically stable llama single-domain antibody fragments for oral immunotherapy. Appl. Microbiol. Biotechnol. 2006, 72, 544–551. [Google Scholar] [CrossRef]
- Famm, K.; Hansen, L.; Christ, D.; Winter, G. Thermodynamically stable aggregation-resistant antibody domains through directed evolution. J. Mol. Biol. 2008, 376, 926–931. [Google Scholar] [CrossRef]
- Jespers, L.; Schon, O.; Famm, K.; Winter, G. Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat. Biotechnol. 2004, 22, 1161–1165. [Google Scholar]
- Stijlemans, B.; Conrath, K.; Cortez-Retamozo, V.; Van Xong, H.; Wyns, L.; Senter, P.; Revets, H.; De Baetselier, P.; Muyldermans, S.; Magez, S. Efficient targeting of conserved cryptic epitopes of infectious agents by single domain antibodies. African trypanosomes as paradigm. J. Biol. Chem. 2004, 279, 1256–1261. [Google Scholar] [PubMed]
- De Genst, E.; Silence, K.; Decanniere, K.; Conrath, K.; Loris, R.; Kinne, J.; Muyldermans, S.; Wyns, L. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. USA 2006, 103, 4586–4591. [Google Scholar]
- Stanfield, R.L.; Dooley, H.; Flajnik, M.F.; Wilson, I.A. Crystal structure of the shark single-domain antibody V region in complex with lysozyme. Science 2004, 305, 1770–1773. [Google Scholar]
- Desmyter, A.; Transue, T.R.; Ghahroudi, M.A.; Thi, M.H.; Poortmans, F.; Hamers, R.; Muyldermans, S.; Wyns, L. Crystal structure of a camel single-domain VH antibody fragment in complex with lysozyme. Nat. Struct. Biol. 1996, 3, 803–811. [Google Scholar]
- Hmila, I.; Abdallah, R.B.A.; Saerens, D.; Benlasfar, Z.; Conrath, K.; Ayeb, M.E.; Muyldermans, S.; Bouhaouala-Zahar, B. VHH, bivalent domains and chimeric heavy chain-only antibodies with high neutralizing efficacy for scorpion toxin AahI’. Mol. Immunol. 2008, 45, 3847–3856. [Google Scholar]
- Harmsen, M.M.; van Solt, C.B.; Fijten, H.P. Enhancement of toxin- and virus-neutralizing capacity of single-domain antibody fragments by N-glycosylation. Appl. Microbiol. Biotechnol. 2009, 84, 1087–1094. [Google Scholar] [CrossRef]
- Harmsen, M.M.; Fijten, H.P.; Engel, B.; Dekker, A.; Eblé, P.L. Passive immunization llama single-domain antibody fragments reduces foot-and-mouth disease transmission between pigs. Vaccine 2009, 27, 1904–1911. [Google Scholar]
- Koch-Nolte, F.; Reyelt, J.; Schössow, B.; Schwarz, N.; Scheuplein, F.; Rothenburg, S.; Haag, F.; Alzogaray, V.; Cauerhff, A.; Goldbaum, F.A. Single domain antibodies from llama effectively and specifically block T cell ecto-ADP-ribosyltransferase ART2.2 in vivo. FASEB J. 2007, 21, 3490–3498. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.; Hirama, T.; Chen, W.; Soltyk, A.L.; Brunton, J.; MacKenzie, C.R.; Zhang, J. A novel pentamer versus pentamer approach to generating neutralizers of verotoxin 1. Mol. Immunol. 2007, 44, 2487–2491. [Google Scholar]
- Forsman, A.; Beirnaert, E.; Aasa-Chapman, M.M.; Hoorelbeke, B.; Hijazi, K.; Koh, W.; Tack, V.; Szynol, A.; Kelly, C.; McKnight, A.; Verrips, T.; de Haard, H.; Weiss, R.A. Llama antibody fragments with cross-subtype human immunodeficiency virus type 1 (HIV-1)-neutralizing properties and high affinity for HIV-1 gp120. J. Virol. 2008, 82, 12069–12081. [Google Scholar]
- Garaicoechea, L.; Olichon, A.; Marcoppido, G.; Wigdorovitz, A.; Mozgovoj, M.; Saif, L.; Surrey, T.; Parreño, V. Llama-derived single-chain antibody fragments directed to rotavirus VP6 protein possess broad neutralizing activity in vitro and confer protection against diarrhea in mice. J. Virol. 2008, 82, 9753–9764. [Google Scholar]
- van der Vaart, J.M.; Pant, N.; Wolvers, D.; Bezemer, S.; Hermans, P.W.; Bellamy, K.; Sarker, S.A.; van der Logt, C.P.; Svensson, L.; Verrips, C.T.; Hammarstrom, L.; van Klinken, B.J. Reduction in morbidity of rotavirus induced diarrhoea in mice by yeast produced monovalent llama-derived antibody fragments. Vaccine 2006, 24, 4130–4137. [Google Scholar]
- Koide, A.; Tereshko, V.; Uysal, S.; Margalef, K.; Kossiakoff, A.A.; Koide, S. Exploring the capacity of minimalist protein interfaces: interface energetics and affinity maturation to picomolar KD of a single-domain antibody with a flat paratope. J. Mol. Biol. 2007, 373, 941–953. [Google Scholar]
- Cortez-Retamozo, V.; Backmann, N.; Senter, P.D.; Wernery, U.; De Baetselier, P.; Muyldermans, S.; Revets, H. Efficient cancer therapy with a nanobody-based conjugate. Cancer Res. 2004, 64, 2853–2857. [Google Scholar]
- Saerens, D.; Kinne, J.; Bosmans, E.; Wernery, U.; Muyldermans, S.; Conrath, K. Single domain antibodies derived from dromedary lymph node and peripheral blood lymphocytes sensing conformational variants of prostate-specific antigen. J. Biol. Chem. 2004, 279, 51965–51972. [Google Scholar] [PubMed]
- Deckers, N.; Saerens, D.; Kanobana, K.; Conrath, K.; Victor, B.; Wernery, U.; Vercruysse, J.; Muyldermans, S.; Dorny, P. anobodies, a promising tool for species-specific diagnosis of Taenia solium cysticercosis. Int. J. Parasitol. 2009, 39, 625–633. [Google Scholar]
- Li, S.; Zheng, W.; Kuolee, R.; Hirama, T.; Henry, M.; Makvandi-Nejad, S.; Fjällman, T.; Chen, W.; Zhang, J. Pentabody-mediated antigen delivery induces antigen-specific mucosal immune response. Mol. Immunol. 2009, 46, 1718–1726. [Google Scholar] [PubMed]
- Ben Abderrazek, R.; Hmila, I.; Vincke, C.; Benlasfar, Z.; Pellis, M.; Dabbek, H.; Saerens, D.; El Ayeb, M.; Muyldermans, S.; Bouhaouala-Zahar, B. Identification of potent nanobodies to neutralize the most poisonous polypeptide from scorpion venom. Biochem. J. 2009, 424, 263–272. [Google Scholar]
- Hmila, I.; Saerens, D.; Ben Abderrazek, R.; Vincke, C.; Abidi, N.; Benlasfar, Z.; Govaert, J.; El Ayeb, M.; Bouhaouala-Zahar, B.; Muyldermans, S. A bispecific antibody to provide full protection against lethal scorpion envenoming. FASEB J. 2010. Epub ahead of print. [Google Scholar]
- Demarest, S.J.; Hariharan, M.; Elia, M.; Salbato, J.; Jin, P.; Bird, C.; Short, J.M.; Kimmel, B.E.; Dudley, M.; Woodnutt, G.; Hansen, G. Neutralization of Clostridium difficile toxin A using antibody combinations. MAbs 2010, 2. Epub ahead of print. [Google Scholar]
- Nowakowski, A.; Wang, C.; Powers, D.B.; Amersdorfer, P.; Smith, T.J.; Montgomery, V.A.; Sheridan, R.; Blake, R.; Smith, L.A.; Marks, J.D. Potent neutralization of botulinum neurotoxin by recombinant oligoclonal antibody. Proc. Natl. Acad. Sci. USA 2002, 99, 11346–11350. [Google Scholar]
- Pant, N.; Hultberg, A.; Zhao, Y.; Svensson, L.; Pan-Hammarstrom, Q.; Johansen, K.; Pouwels, P.H.; Ruggeri, F.M.; Hermans, P.; Frenken, L.; Boren, T.; Marcotte, H.; Hammarstrom, L. Lactobacilli expressing variable domain of llama heavy-chain antibody fragments (lactobodies) confer protection against rotavirus-induced diarrhea. J. Infect. Dis. 2006, 194, 1580–1588. [Google Scholar]
- Krüger, C.; Hu, Y.; Pan, Q.; Marcotte, H.; Hultberg, A.; Delwar, D.; van Dalen, P.J.; Pouwels, P.H.; Leer, R.J.; Kelly, C.G.; van Dollenweerd, C.; Ma, J.K.; Hammarström, L. In situ delivery of passive immunity by lactobacilli producing single-chain antibodies. Nat. Biotechnol. 2002, 20, 702–706. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hussack, G.; Tanha, J. Toxin-Specific Antibodies for the Treatment of Clostridium difficile: Current Status and Future Perspectives. Toxins 2010, 2, 998-1018. https://doi.org/10.3390/toxins2050998
Hussack G, Tanha J. Toxin-Specific Antibodies for the Treatment of Clostridium difficile: Current Status and Future Perspectives. Toxins. 2010; 2(5):998-1018. https://doi.org/10.3390/toxins2050998
Chicago/Turabian StyleHussack, Greg, and Jamshid Tanha. 2010. "Toxin-Specific Antibodies for the Treatment of Clostridium difficile: Current Status and Future Perspectives" Toxins 2, no. 5: 998-1018. https://doi.org/10.3390/toxins2050998
APA StyleHussack, G., & Tanha, J. (2010). Toxin-Specific Antibodies for the Treatment of Clostridium difficile: Current Status and Future Perspectives. Toxins, 2(5), 998-1018. https://doi.org/10.3390/toxins2050998