Heat-Labile Enterotoxin: Beyond G M1 Binding

Abstract
:
1. Introduction
1.1. Enterotoxigenic Escherichia coli
1.2. Enterotoxins produced by ETEC

1.3. Catalytic activity of LT
1.4. Homology to CT
2. Regulation of LT Production
2.1. Growth conditions inducing the release of LT
2.2. Regulation by H-NS
2.3. Feedback from cAMP

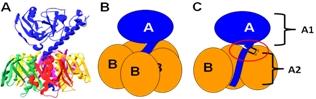
3. Holotoxin Assembly
3.1. Periplasmic localization
3.2. Factors influencing oligomerization
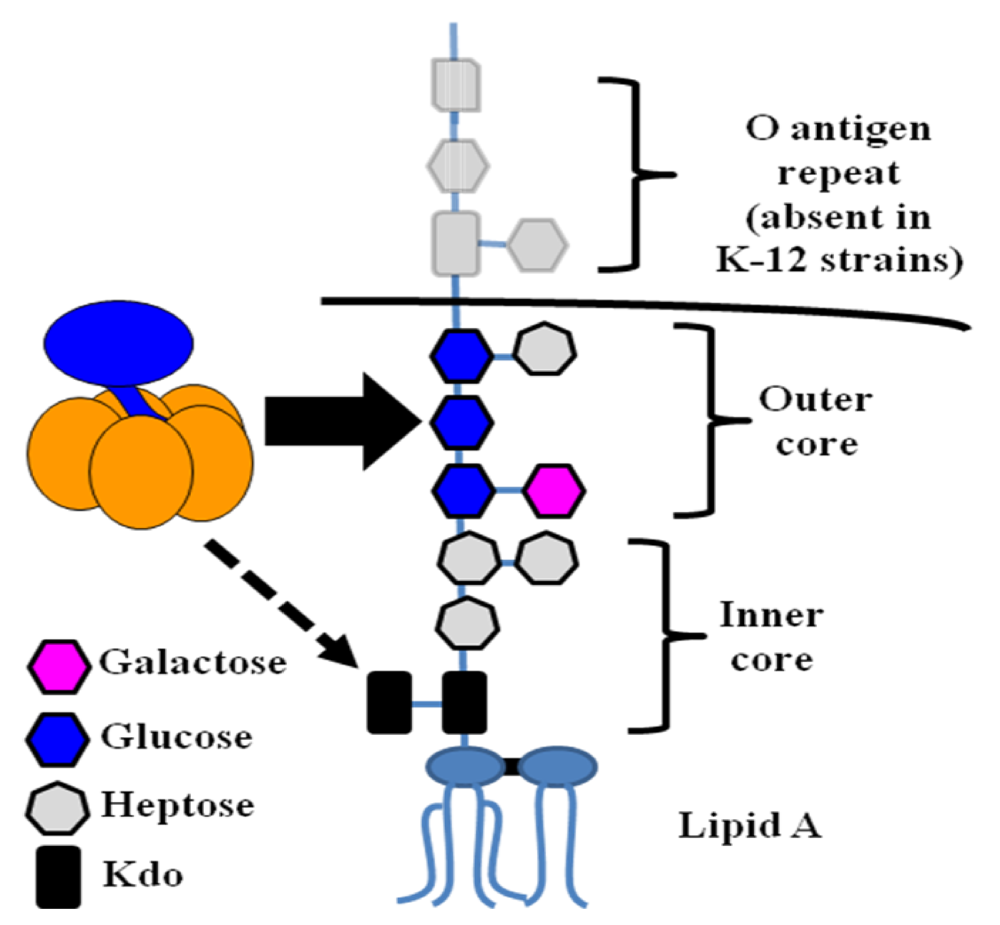
4. Secretion
4.1. Type II secretion
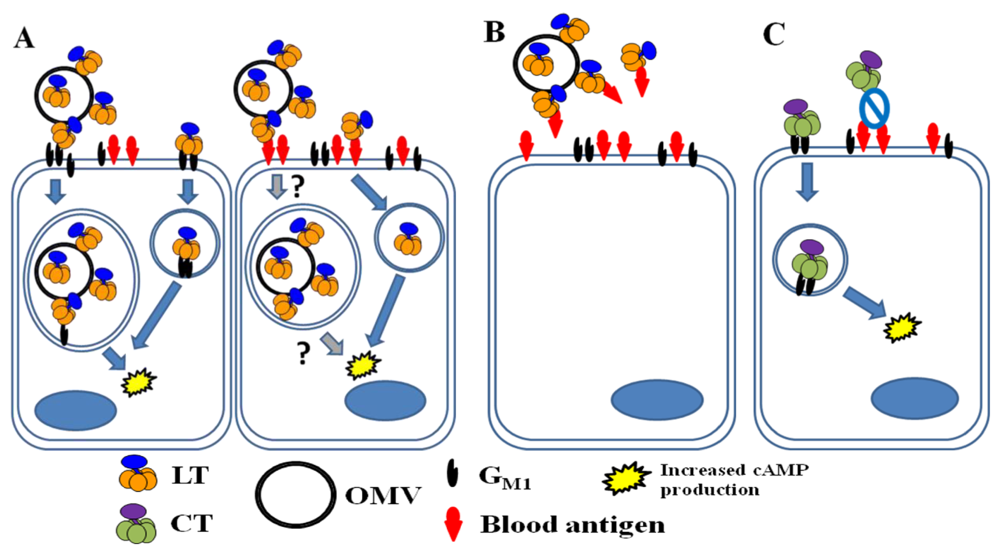
4.2. Secretion via outer membrane vesicles
5. Ganglioside Binding
5.1. LT’s interaction with GM1

5.2. Additional non-GM1 substrates

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ganglioside | Structure | LTB binding | CTB binding |
|---|---|---|---|
| GM1 |  | ++++ | ++++ |
| Asialo-GM1 |  | + | - |
| Paragloboside |  | + | - |
| GD1b |  | + | +/- |
| GM2 |  | +/- | +/- |
6. Blood Sugar Binding
6.1. LTB binds to A-type blood sugars
6.2. Residues of LTB involved in blood sugar binding

6.3. ETEC infection and blood type
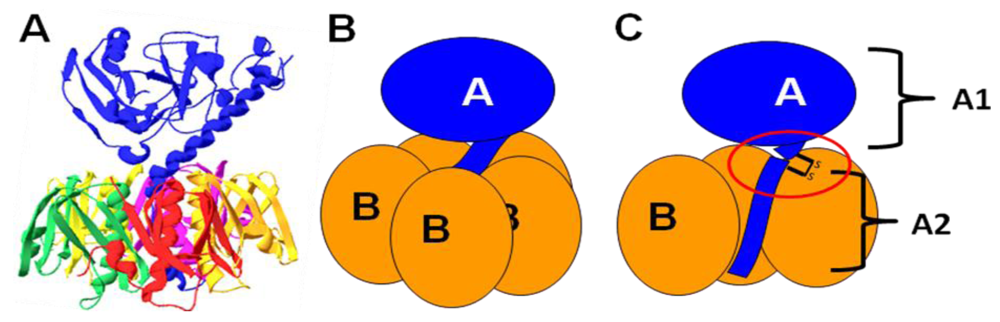
7. Lipopolysaccharide Binding
7.1. Binding of LT to the E. coli surface is due to an association with lipopolysaccharide

7.2. LTB residues involved in LPS binding
7.3. Possible roles of LPS binding
8. Conclusions

Acknowledgements
References
- Coster, T.S.; Wolf, M.K.; Hall, E.R.; Cassels, F.J.; Taylor, D.N.; Liu, C.T.; Trespalacios, F.C.; DeLorimier, A.; Angleberger, D.R.; McQueen, C.E. Immune response, ciprofloxacin activity, and gender differences after human experimental challenge by two strains of enterotoxigenic Escherichia coli. Infect. Immun. 2007, 75, 252–259. [Google Scholar] [PubMed]
- Vicente, A.C.; Teixeira, L.F.; Iniguez-Rojas, L.; Luna, M.G.; Silva, L.; Andrade, J.R.; Guth, B.E. Outbreaks of cholera-like diarrhoea caused by enterotoxigenic Escherichia coli in the Brazilian Amazon Rainforest. Trans. R. Soc. Trop. Med. Hyg. 2005, 99, 669–674. [Google Scholar]
- Sack, R.B.; Gorbach, S.L.; Banwell, J.G.; Jacobs, B.; Chatterjee, B.D.; Mitra, R.C. Enterotoxigenic Escherichia coli isolated from patients with severe cholera-like disease. J. Infect. Dis. 1971, 123, 378–385. [Google Scholar]
- Qadri, F.; Svennerholm, A.M.; Faruque, A.S.; Sack, R.B. Enterotoxigenic Escherichia coli in developing countries: epidemiology, microbiology, clinical features, treatment, and prevention. Clin. Microbiol. Rev. 2005, 18, 465–483. [Google Scholar]
- Fleckenstein, J.M.; Hardwidge, P.R.; Munson, G.P.; Rasko, D.A.; Sommerfelt, H.; Steinsland, H. Molecular Mechanisms of Enterotoxigenic Escherichia coli Infection. Microbes Infect. 2010, 12, 89–98. [Google Scholar]
- Sanchez, J.; Holmgren, J. Virulence factors, pathogenesis and vaccine protection in cholera and ETEC diarrhea. Curr. Opin. Immunol. 2005, 17, 388–398. [Google Scholar]
- Johnson, T.J.; Nolan, L.K. Pathogenomics of the virulence plasmids of Escherichia coli. Microbiol. Mol. Biol. Rev. 2009, 73, 750–774. [Google Scholar]
- Svennerholm, A.M.; Tobias, J. Vaccines against enterotoxigenic Escherichia coli. Expert Rev. Vaccines 2008, 7, 795–804. [Google Scholar]
- Wolf, M.K. Occurrence, distribution, and associations of O and H serogroups, colonization factor antigens, and toxins of enterotoxigenic Escherichia coli. Clin. Microbiol. Rev. 1997, 10, 569–584. [Google Scholar] [PubMed]
- Hughes, J.M.; Murad, F.; Chang, B.; Guerrant, R.L. Role of cyclic GMP in the action of heat-stable enterotoxin of Escherichia coli. Nature 1978, 271, 755–756. [Google Scholar]
- Connell, T.D.; Holmes, R.K. Characterization of hybrid toxins produced in Escherichia coli by assembly of A and B polypeptides from type I and type II heat-labile enterotoxins. Infect. Immun. 1992, 60, 1653–1661. [Google Scholar]
- Gill, D.M.; Clements, J.D.; Robertson, D.C.; Finkelstein, R.A. Subunit number and arrangement in Escherichia coli heat-labile enterotoxin. Infect. Immun. 1981, 33, 677–682. [Google Scholar]
- Johnson, A.M.; Kaushik, R.S.; Francis, D.H.; Fleckenstein, J.M.; Hardwidge, P.R. Heat-labile enterotoxin promotes Escherichia coli adherence to intestinal epithelial cells. J. Bacteriol. 2009, 191, 178–186. [Google Scholar]
- Allen, K.P.; Randolph, M.M.; Fleckenstein, J.M. Importance of heat-labile enterotoxin in colonization of the adult mouse small intestine by human enterotoxigenic Escherichia coli strains. Infect. Immun. 2006, 74, 869–875. [Google Scholar]
- Berberov, E.M.; Zhou, Y.; Francis, D.H.; Scott, M.A.; Kachman, S.D.; Moxley, R.A. Relative importance of heat-labile enterotoxin in the causation of severe diarrheal disease in the gnotobiotic piglet model by a strain of enterotoxigenic Escherichia coli that produces multiple enterotoxins. Infect. Immun. 2004, 72, 3914–3924. [Google Scholar]
- Hardy, S.J.; Holmgren, J.; Johansson, S.; Sanchez, J.; Hirst, T.R. Coordinated assembly of multisubunit proteins: oligomerization of bacterial enterotoxins in vivo and in vitro. Proc. Natl. Acad. Sci. USA 1988, 85, 7109–7113. [Google Scholar]
- Spangler, B.D. Structure and function of cholera toxin and the related Escherichia coli heat-labile enterotoxin. Microbiol. Rev. 1992, 56, 622–647. [Google Scholar]
- Moss, J.; Richardson, S.H. Activation of adenylate cyclase by heat-labile Escherichia coli enterotoxin. Evidence for ADP-ribosyltransferase activity similar to that of choleragen. J. Clin. Invest. 1978, 62, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Sears, C.L.; Kaper, J.B. Enteric bacterial toxins: mechanisms of action and linkage to intestinal secretion. Microbiol. Rev. 1996, 60, 167–215. [Google Scholar]
- Chakraborty, K.; Ghosh, S.; Koley, H.; Mukhopadhyay, A.K.; Ramamurthy, T.; Saha, D.R.; Mukhopadhyay, D.; Roychowdhury, S.; Hamabata, T.; Takeda, Y.; Das, S. Bacterial exotoxins downregulate cathelicidin (hCAP-18/LL-37) and human beta-defensin 1 (HBD-1) expression in the intestinal epithelial cells. Cell. Microbiol. 2008, 10, 2520–2537. [Google Scholar]
- Grant, C.C.; Messer, R.J.; Cieplak, W., Jr. Role of trypsin-like cleavage at arginine 192 in the enzymatic and cytotonic activities of Escherichia coli heat-labile enterotoxin. Infect. Immun. 1994, 62, 4270–4278. [Google Scholar]
- Clements, J.D.; Finkelstein, R.A. Isolation and characterization of homogeneous heat-labile enterotoxins with high specific activity from Escherichia coli cultures. Infect. Immun. 1979, 24, 760–769. [Google Scholar]
- Lencer, W.I.; Hirst, T.R.; Holmes, R.K. Membrane traffic and the cellular uptake of cholera toxin. Biochim. Biophys. Acta 1999, 1450, 177–190. [Google Scholar]
- Okamoto, K.; Nomura, T.; Fujii, Y.; Yamanaka, H. Contribution of the disulfide bond of the A subunit to the action of Escherichia coli heat-labile enterotoxin. J. Bacteriol. 1998, 180, 1368–1374. [Google Scholar]
- Yamamoto, T.; Tamura, T.; Yokota, T.; Takano, T. Overlapping genes in the heat-labile enterotoxin operon originating from Escherichia coli human strain. Mol. Gen. Genet. 1982, 188, 356–359. [Google Scholar]
- Sandkvist, M.; Hirst, T.R.; Bagdasarian, M. Alterations at the carboxyl terminus change assembly and secretion properties of the B subunit of Escherichia coli heat-labile enterotoxin. J. Bacteriol. 1987, 169, 4570–4576. [Google Scholar]
- Yamamoto, T.; Nakazawa, T.; Miyata, T.; Kaji, A.; Yokota, T. Evolution and structure of two ADP-ribosylation enterotoxins, Escherichia coli heat-labile toxin and cholera toxin. FEBS Lett. 1984, 169, 241–246. [Google Scholar]
- Holmner, A.; Askarieh, G.; Okvist, M.; Krengel, U. Blood group antigen recognition by Escherichia coli heat-labile enterotoxin. J. Mol. Biol. 2007, 371, 754–764. [Google Scholar]
- Yamamoto, T.; Gojobori, T.; Yokota, T. Evolutionary origin of pathogenic determinants in enterotoxigenic Escherichia coli and Vibrio cholerae O1. J. Bacteriol. 1987, 169, 1352–1357. [Google Scholar]
- Schlor, S.; Riedl, S.; Blass, J.; Reidl, J. Genetic rearrangements of the regions adjacent to genes encoding heat-labile enterotoxins (eltAB) of enterotoxigenic Escherichia coli strains. Appl. Environ. Microbiol. 2000, 66, 352–358. [Google Scholar]
- Scotland, S.M.; Day, N.P.; Rowe, B. Acquisition and maintenance of enterotoxin plasmids in wild-type strains of Escherichia coli. J. Gen. Microbiol. 1983, 129, 3111–3120. [Google Scholar]
- Krukonis, E.S.; Yu, R.R.; Dirita, V.J. The Vibrio cholerae ToxR/TcpP/ToxT virulence cascade: distinct roles for two membrane-localized transcriptional activators on a single promoter. Mol. Microbiol. 2000, 38, 67–84. [Google Scholar]
- Kunkel, S.L.; Robertson, D.C. Factors affecting release of heat-labile enterotoxin by enterotoxigenic Escherichia coli. Infect. Immun. 1979, 23, 652–659. [Google Scholar]
- Mundell, D.H.; Anselmo, C.R.; Wishnow, R.M. Factors influencing heat-labile Escherichia coli enterotoxin activity. Infect. Immun. 1976, 14, 383–388. [Google Scholar]
- Gibert, I.; Barbe, J. Cyclic AMP stimulates transcription of the structural gene of the outer-membrane protein OmpA of Escherichia coli. FEMS Microbiol. Lett. 1990, 56, 307–311. [Google Scholar]
- Trachman, J.D.; Yasmin, M. Thermo-osmoregulation of heat-labile enterotoxin expression by Escherichia coli. Curr. Microbiol. 2004, 49, 353–360. [Google Scholar]
- Takashi, K.; Fujita, I.; Kobari, K. Effects of short chain fatty acids on the production of heat-labile enterotoxin from enterotoxigenic Escherichia coli. Jpn. J. Pharmacol. 1989, 50, 495–498. [Google Scholar]
- Binder, H.J. Role of colonic short-chain fatty acid transport in diarrhea. Annu. Rev. Physiol. 2010, 72, 297–313. [Google Scholar]
- Lasaro, M.A.; Rodrigues, J.F.; Mathias-Santos, C.; Guth, B.E.; Regua-Mangia, A.; Piantino Ferreira, A.J.; Takagi, M.; Cabrera-Crespo, J.; Sbrogio-Almeida, M.E.; de Souza Ferreira, L.C. Production and release of heat-labile toxin by wild-type human-derived enterotoxigenic Escherichia coli. FEMS Immunol. Med. Microbiol. 2006, 48, 123–131. [Google Scholar]
- Sjoling, A.; Qadri, F.; Nicklasson, M.; Begum, Y.A.; Wiklund, G.; Svennerholm, A.M. In vivo expression of the heat stable (estA) and heat labile (eltB) toxin genes of enterotoxigenic Escherichia coli (ETEC). Microbes Infect. 2006, 8, 2797–2802. [Google Scholar]
- Dallas, W.S.; Falkow, S. Amino acid sequence homology between cholera toxin and Escherichia coli heat-labile toxin. Nature 1980, 288, 499–501. [Google Scholar]
- Trachman, J.D.; Maas, W.K. Temperature regulation of heat-labile enterotoxin (LT) synthesis in Escherichia coli is mediated by an interaction of H-NS protein with the LT A-subunit DNA. J. Bacteriol. 1998, 180, 3715–3718. [Google Scholar]
- Yang, J.; Tauschek, M.; Strugnell, R.; Robins-Browne, R.M. The H-NS protein represses transcription of the eltAB operon, which encodes heat-labile enterotoxin in enterotoxigenic Escherichia coli, by binding to regions downstream of the promoter. Microbiology 2005, 151, 1199–1208. [Google Scholar]
- Hamilton, D.L.; Johnson, M.R.; Forsyth, G.W.; Roe, W.E.; Nielsen, N.O. The effect of cholera toxin and heat labile and heat stable Escherichia coli enterotoxin on cyclic AMP concentrations in small intestinal mucosa of pig and rabbit. Can. J. Comp. Med. 1978, 42, 327–331. [Google Scholar]
- Bodero, M.D.; Munson, G.P. Cyclic AMP receptor protein-dependent repression of heat-labile enterotoxin. Infect. Immun. 2009, 77, 791–798. [Google Scholar]
- Ferraris, R.P.; Yasharpour, S.; Lloyd, K.C.; Mirzayan, R.; Diamond, J.M. Luminal glucose concentrations in the gut under normal conditions. Am. J. Physiol. 1990, 259, G822–G837. [Google Scholar]
- Spicer, E.K.; Noble, J.A. Escherichia coli heat-labile enterotoxin. Nucleotide sequence of the A subunit gene. J. Biol. Chem. 1982, 257, 5716–5721. [Google Scholar]
- Yamamoto, T.; Tamura, T.; Ryoji, M.; Kaji, A.; Yokota, T.; Takano, T. Sequence analysis of the heat-labile enterotoxin subunit B gene originating in human enterotoxigenic Escherichia coli. J. Bacteriol. 1982, 152, 506–509. [Google Scholar]
- Hirst, T.R.; Randall, L.L.; Hardy, S.J. Cellular location of heat-labile enterotoxin in Escherichia coli. J. Bacteriol. 1984, 157, 637–642. [Google Scholar]
- Hofstra, H.; Witholt, B. Heat-labile enterotoxin in Escherichia coli. Kinetics of association of subunits into periplasmic holotoxin. J. Biol. Chem. 1985, 260, 16037–16044. [Google Scholar] [PubMed]
- Chung, W.Y.; Carter, R.; Hardy, T.; Sack, M.; Hirst, T.R.; James, R.F. Inhibition of Escherichia coli heat-labile enterotoxin B subunit pentamer (EtxB5) assembly in vitro using monoclonal antibodies. J. Biol. Chem. 2006, 281, 39465–39470. [Google Scholar]
- Yu, J.; Webb, H.; Hirst, T.R. A homologue of the Escherichia coli DsbA protein involved in disulphide bond formation is required for enterotoxin biogenesis in Vibrio cholerae. Mol. Microbiol. 1992, 6, 1949–1958. [Google Scholar]
- Ruddock, L.W.; Webb, H.M.; Ruston, S.P.; Cheesman, C.; Freedman, R.B.; Hirst, T.R. A pH-dependent conformational change in the B-subunit pentamer of Escherichia coli heat-labile enterotoxin: structural basis and possible functional role for a conserved feature of the AB5 toxin family. Biochemistry 1996, 35, 16069–16076. [Google Scholar]
- Streatfield, S.J.; Sandkvist, M.; Sixma, T.K.; Bagdasarian, M.; Hol, W.G.; Hirst, T.R. Intermolecular interactions between the A and B subunits of heat-labile enterotoxin from Escherichia coli promote holotoxin assembly and stability in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 12140–12144. [Google Scholar]
- Sixma, T.K.; Pronk, S.E.; Kalk, K.H.; Wartna, E.S.; van Zanten, B.A.; Witholt, B.; Hol, W.G. Crystal structure of a cholera toxin-related heat-labile enterotoxin from E. coli. Nature 1991, 351, 371–377. [Google Scholar] [PubMed]
- Gankema, H.; Wensink, J.; Guinee, P.A.; Jansen, W.H.; Witholt, B. Some characteristics of the outer membrane material released by growing enterotoxigenic Escherichia coli. Infect. Immun. 1980, 29, 704–713. [Google Scholar]
- Jacks, T.M.; Wu, B.J.; Braemer, A.C.; Bidlack, D.E. Properties of the enterotoxic component in Escherichia coli enteropathogenic for swine. Infect. Immun. 1973, 7, 178–189. [Google Scholar]
- Neill, R.J.; Ivins, B.E.; Holmes, R.K. Synthesis and secretion of the plasmid-coded heat-labile enterotoxin of Escherichia coli in Vibrio cholerae. Science 1983, 221, 289–291. [Google Scholar]
- Sandkvist, M.; Michel, L.O.; Hough, L.P.; Morales, V.M.; Bagdasarian, M.; Koomey, M.; DiRita, V.J. General secretion pathway (eps) genes required for toxin secretion and outer membrane biogenesis in Vibrio cholerae. J. Bacteriol. 1997, 179, 6994–7003. [Google Scholar]
- Johnson, T.L.; Abendroth, J.; Hol, W.G.; Sandkvist, M. Type II secretion: from structure to function. FEMS Microbiol. Lett. 2006, 255, 175–186. [Google Scholar]
- Tauschek, M.; Gorrell, R.J.; Strugnell, R.A.; Robins-Browne, R.M. Identification of a protein secretory pathway for the secretion of heat-labile enterotoxin by an enterotoxigenic strain of Escherichia coli. Proc. Natl. Acad. Sci. USA 2002, 99, 7066–7071. [Google Scholar]
- Horstman, A.L.; Kuehn, M.J. Bacterial surface association of heat-labile enterotoxin through lipopolysaccharide after secretion via the general secretory pathway. J. Biol. Chem. 2002, 277, 32538–32545. [Google Scholar]
- Yang, J.; Baldi, D.L.; Tauschek, M.; Strugnell, R.A.; Robins-Browne, R.M. Transcriptional regulation of the yghJ-pppA-yghG-gspCDEFGHIJKLM cluster, encoding the type II secretion pathway in enterotoxigenic Escherichia coli. J. Bacteriol. 2007, 189, 142–150. [Google Scholar]
- Francetic, O.; Belin, D.; Badaut, C.; Pugsley, A.P. Expression of the endogenous type II secretion pathway in Escherichia coli leads to chitinase secretion. EMBO J. 2000, 19, 6697–6703. [Google Scholar]
- Mudrak, B.; Kuehn, M.J. Specificity of the type II secretion systems of enterotoxigenic Escherichia coli and Vibrio cholerae for heat-labile enterotoxin and cholera toxin. J. Bacteriol. 2010. [Google Scholar]
- Hirst, T.R.; Sanchez, J.; Kaper, J.B.; Hardy, S.J.; Holmgren, J. Mechanism of toxin secretion by Vibrio cholerae investigated in strains harboring plasmids that encode heat-labile enterotoxins of Escherichia coli. Proc. Natl. Acad. Sci. USA 1984, 81, 7752–7756. [Google Scholar]
- Connell, T.D.; Metzger, D.J.; Wang, M.; Jobling, M.G.; Holmes, R.K. Initial studies of the structural signal for extracellular transport of cholera toxin and other proteins recognized by Vibrio cholerae. Infect. Immun. 1995, 63, 4091–4098. [Google Scholar]
- Brown, E.A.; Hardwidge, P.R. Biochemical characterization of the enterotoxigenic Escherichia coli LeoA protein. Microbiology 2007, 153, 3776–3784. [Google Scholar]
- Fleckenstein, J.M.; Lindler, L.E.; Elsinghorst, E.A.; Dale, J.B. Identification of a gene within a pathogenicity island of enterotoxigenic Escherichia coli H10407 required for maximal secretion of the heat-labile enterotoxin. Infect. Immun. 2000, 68, 2766–2774. [Google Scholar]
- Turner, S.M.; Chaudhuri, R.R.; Jiang, Z.D.; DuPont, H.; Gyles, C.; Penn, C.W.; Pallen, M.J.; Henderson, I.R. Phylogenetic comparisons reveal multiple acquisitions of the toxin genes by enterotoxigenic Escherichia coli strains of different evolutionary lineages. J. Clin. Microbiol. 2006, 44, 4528–4536. [Google Scholar]
- Horstman, A.L.; Kuehn, M.J. Enterotoxigenic Escherichia coli secretes active heat-labile enterotoxin via outer membrane vesicles. J. Biol. Chem. 2000, 275, 12489–12496. [Google Scholar]
- Wai, S.N.; Takade, A.; Amako, K. The release of outer membrane vesicles from the strains of enterotoxigenic Escherichia coli. Microbiol. Immunol. 1995, 39, 451–456. [Google Scholar]
- Kuehn, M.J.; Kesty, N.C. Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev. 2005, 19, 2645–2655. [Google Scholar]
- Ellis, T.N.; Kuehn, M.J. Virulence and immunomodulatory roles of bacterial outer membrane vesicles. Microbiol. Mol. Biol. Rev. 2010, 74, 81–94. [Google Scholar]
- Moss, J.; Osborne, J.C., Jr.; Fishman, P.H.; Nakaya, S.; Robertson, D.C. Escherichia coli heat-labile enterotoxin. Ganglioside specificity and ADP-ribosyltransferase activity. J. Biol. Chem. 1981, 256, 12861–12865. [Google Scholar] [PubMed]
- Merritt, E.A.; Sixma, T.K.; Kalk, K.H.; van Zanten, B.A.; Hol, W.G. Galactose-binding site in Escherichia coli heat-labile enterotoxin (LT) and cholera toxin (CT). Mol. Microbiol. 1994, 13, 745–753. [Google Scholar]
- De Wolf, M.J.; Fridkin, M.; Kohn, L.D. Tryptophan residues of cholera toxin and its A and B protomers. Intrinsic fluorescence and solute quenching upon interacting with the ganglioside GM1, oligo-GM1, or dansylated oligo-GM1. J. Biol. Chem. 1981, 256, 5489–5496. [Google Scholar] [PubMed]
- Ruddock, L.W.; Coen, J.J.; Cheesman, C.; Freedman, R.B.; Hirst, T.R. Assembly of the B subunit pentamer of Escherichia coli heat-labile enterotoxin. Kinetics and molecular basis of rate-limiting steps in vitro. J. Biol. Chem. 1996, 271, 19118–19123. [Google Scholar] [PubMed]
- Merritt, E.A.; Sarfaty, S.; van den Akker, F.; L'Hoir, C.; Martial, J.A.; Hol, W.G. Crystal structure of cholera toxin B-pentamer bound to receptor GM1 pentasaccharide. Protein Sci. 1994, 3, 166–175. [Google Scholar]
- Wolf, A.A.; Jobling, M.G.; Saslowsky, D.E.; Kern, E.; Drake, K.R.; Kenworthy, A.K.; Holmes, R.K.; Lencer, W.I. Attenuated endocytosis and toxicity of a mutant cholera toxin with decreased ability to cluster ganglioside GM1 molecules. Infect. Immun. 2008, 76, 1476–1484. [Google Scholar]
- Holmgren, J.; Fredman, P.; Lindblad, M.; Svennerholm, A.M.; Svennerholm, L. Rabbit intestinal glycoprotein receptor for Escherichia coli heat-labile enterotoxin lacking affinity for cholera toxin. Infect. Immun. 1982, 38, 424–433. [Google Scholar]
- Fan, E.; Merritt, E.A.; Zhang, Z.; Pickens, J.C.; Roach, C.; Ahn, M.; Hol, W.G. Exploration of the GM1 receptor-binding site of heat-labile enterotoxin and cholera toxin by phenyl-ring-containing galactose derivatives. Acta Crystallogr. D Biol. Crystallogr. 2001, 57, 201–212. [Google Scholar]
- Minke, W.E.; Roach, C.; Hol, W.G.; Verlinde, C.L. Structure-based exploration of the ganglioside GM1 binding sites of Escherichia coli heat-labile enterotoxin and cholera toxin for the discovery of receptor antagonists. Biochemistry 1999, 38, 5684–5692. [Google Scholar]
- Wernick, N.L.B.; Chinnapen, D.J.-F.; Cho, J.A.; Lencer, W.I. Cholera toxin: An intracellular journey into the cytosol by way of the endoplasmic reticulum. Toxins 2010, 2, 310–325. [Google Scholar]
- Connell, T.D. Cholera toxin, LT-I, LT-IIa and LT-IIb: the critical role of ganglioside binding in immunomodulation by type I and type II heat-labile enterotoxins. Expert Rev. Vaccines 2007, 6, 821–834. [Google Scholar]
- Mudrak, B.; Rodriguez, D.L.; Kuehn, M.J. Residues of heat-labile enterotoxin involved in bacterial cell surface binding. J. Bacteriol. 2009, 191, 2917–2925. [Google Scholar]
- Jansson, L.; Angstrom, J.; Lebens, M.; Imberty, A.; Varrot, A.; Teneberg, S. Carbohydrate binding specificities and crystal structure of the cholera toxin-like B-subunit from Citrobacter freundii. Biochimie 2010, 92, 482–490. [Google Scholar]
- MacKenzie, C.R.; Hirama, T.; Lee, K.K.; Altman, E.; Young, N.M. Quantitative analysis of bacterial toxin affinity and specificity for glycolipid receptors by surface plasmon resonance. J. Biol. Chem. 1997, 272, 5533–5538. [Google Scholar]
- Teneberg, S.; Hirst, T.R.; Angstrom, J.; Karlsson, K.A. Comparison of the glycolipid-binding specificities of cholera toxin and porcine Escherichia coli heat-labile enterotoxin: identification of a receptor-active non-ganglioside glycolipid for the heat-labile toxin in infant rabbit small intestine. Glycoconj. J. 1994, 11, 533–540. [Google Scholar]
- Fukuta, S.; Magnani, J.L.; Twiddy, E.M.; Holmes, R.K.; Ginsburg, V. Comparison of the carbohydrate-binding specificities of cholera toxin and Escherichia coli heat-labile enterotoxins LTh-I, LT-IIa, and LT-IIb. Infect. Immun. 1988, 56, 1748–1753. [Google Scholar]
- Chatterjee, A.; Chowdhury, R. Bile and unsaturated fatty acids inhibit the binding of cholera toxin and Escherichia coli heat-labile enterotoxin to GM1 receptor. Antimicrob. Agents Chemother. 2008, 52, 220–224. [Google Scholar]
- Lasaro, M.A.; Rodrigues, J.F.; Mathias-Santos, C.; Guth, B.E.; Balan, A.; Sbrogio-Almeida, M.E.; Ferreira, L.C. Genetic diversity of heat-labile toxin expressed by enterotoxigenic Escherichia coli strains isolated from humans. J. Bacteriol. 2008, 190, 2400–2410. [Google Scholar]
- Balanzino, L.E.; Barra, J.L.; Galvan, E.M.; Roth, G.A.; Monferran, C.G. Interaction of cholera toxin and Escherichia coli heat-labile enterotoxin with glycoconjugates from rabbit intestinal brush border membranes: relationship with ABH blood group determinants. Mol. Cell. Biochem. 1999, 194, 53–62. [Google Scholar]
- Barra, J.L.; Monferran, C.G.; Balanzino, L.E.; Cumar, F.A. Escherichia coli heat-labile enterotoxin preferentially interacts with blood group A-active glycolipids from pig intestinal mucosa and A- and B-active glycolipids from human red cells compared to H-active glycolipids. Mol. Cell. Biochem. 1992, 115, 63–70. [Google Scholar]
- Galvan, E.M.; Roth, G.A.; Monferran, C.G. Participation of ABH glycoconjugates in the secretory response to Escherichia coli heat-labile toxin in rabbit intestine. J. Infect. Dis. 1999, 180, 419–425. [Google Scholar]
- Galvan, E.M.; Diema, C.D.; Roth, G.A.; Monferran, C.G. Ability of blood group A-active glycosphingolipids to act as Escherichia coli heat-labile enterotoxin receptors in HT-29 cells. J. Infect. Dis. 2004, 189, 1556–1564. [Google Scholar]
- Galvan, E.M.; Roth, G.A.; Monferran, C.G. Functional interaction of Escherichia coli heat-labile enterotoxin with blood group A-active glycoconjugates from differentiated HT29 cells. FEBS J. 2006, 273, 3444–3453. [Google Scholar]
- Holmner, A.; Lebens, M.; Teneberg, S.; Angstrom, J.; Okvist, M.; Krengel, U. Novel binding site identified in a hybrid between cholera toxin and heat-labile enterotoxin: 1.9 A crystal structure reveals the details. Structure 2004, 12, 1655–1667. [Google Scholar] [CrossRef] [PubMed]
- Angstrom, J.; Backstrom, M.; Berntsson, A.; Karlsson, N.; Holmgren, J.; Karlsson, K.A.; Lebens, M.; Teneberg, S. Novel carbohydrate binding site recognizing blood group A and B determinants in a hybrid of cholera toxin and Escherichia coli heat-labile enterotoxin B-subunits. J. Biol. Chem. 2000, 275, 3231–3238. [Google Scholar]
- Jansson, L.; Angstrom, J.; Lebens, M.; Teneberg, S. No direct binding of the heat-labile enterotoxin of Escherichia coli to E. coli lipopolysaccharides. Glycoconj. J. 2009, 27, 171–179. [Google Scholar] [PubMed]
- Harris, J.B.; Khan, A.I.; LaRocque, R.C.; Dorer, D.J.; Chowdhury, F.; Faruque, A.S.; Sack, D.A.; Ryan, E.T.; Qadri, F.; Calderwood, S.B. Blood group, immunity, and risk of infection with Vibrio cholerae in an area of endemicity. Infect. Immun. 2005, 73, 7422–7427. [Google Scholar]
- van Loon, F.P.; Clemens, J.D.; Sack, D.A.; Rao, M.R.; Ahmed, F.; Chowdhury, S.; Harris, J.R.; Ali, M.; Chakraborty, J.; Khan, M.R.; et al. ABO blood groups and the risk of diarrhea due to enterotoxigenic Escherichia coli. J. Infect. Dis. 1991, 163, 1243–1246. [Google Scholar] [CrossRef] [PubMed]
- Qadri, F.; Saha, A.; Ahmed, T.; Al Tarique, A.; Begum, Y.A.; Svennerholm, A.M. Disease burden due to enterotoxigenic Escherichia coli in the first 2 years of life in an urban community in Bangladesh. Infect. Immun. 2007, 75, 3961–3968. [Google Scholar]
- Finne, J.; Breimer, M.E.; Hansson, G.C.; Karlsson, K.A.; Leffler, H.; Vliegenthart, J.F.; van Halbeek, H. Novel polyfucosylated N-linked glycopeptides with blood group A, H, X, and Y determinants from human small intestinal epithelial cells. J. Biol. Chem. 1989, 264, 5720–5735. [Google Scholar]
- D'Adamo, P.J.; Kelly, G.S. Metabolic and immunologic consequences of ABH secretor and Lewis subtype status. Altern. Med. Rev. 2001, 6, 390–405. [Google Scholar]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar]
- Horstman, A.L.; Bauman, S.J.; Kuehn, M.J. Lipopolysaccharide 3-deoxy-D-manno-octulosonic acid (Kdo) core determines bacterial association of secreted toxins. J. Biol. Chem. 2004, 279, 8070–8075. [Google Scholar]
- Kesty, N.C.; Mason, K.M.; Reedy, M.; Miller, S.E.; Kuehn, M.J. Enterotoxigenic Escherichia coli vesicles target toxin delivery into mammalian cells. EMBO J. 2004, 23, 4538–4549. [Google Scholar]
- Amor, K.; Heinrichs, D.E.; Frirdich, E.; Ziebell, K.; Johnson, R.P.; Whitfield, C. Distribution of core oligosaccharide types in lipopolysaccharides from Escherichia coli. Infect. Immun. 2000, 68, 1116–1124. [Google Scholar]
- Cox, A.D.; Brisson, J.R.; Varma, V.; Perry, M.B. Structural analysis of the lipopolysaccharide from Vibrio cholerae O139. Carbohydr. Res. 1996, 290, 43–58. [Google Scholar]
- Sixma, T.K.; Kalk, K.H.; van Zanten, B.A.; Dauter, Z.; Kingma, J.; Witholt, B.; Hol, W.G. Refined structure of Escherichia coli heat-labile enterotoxin, a close relative of cholera toxin. J. Mol. Biol. 1993, 230, 890–918. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mudrak, B.; Kuehn, M.J. Heat-Labile Enterotoxin: Beyond G M1 Binding. Toxins 2010, 2, 1445-1470. https://doi.org/10.3390/toxins2061445
Mudrak B, Kuehn MJ. Heat-Labile Enterotoxin: Beyond G M1 Binding. Toxins. 2010; 2(6):1445-1470. https://doi.org/10.3390/toxins2061445
Chicago/Turabian StyleMudrak, Benjamin, and Meta J. Kuehn. 2010. "Heat-Labile Enterotoxin: Beyond G M1 Binding" Toxins 2, no. 6: 1445-1470. https://doi.org/10.3390/toxins2061445
APA StyleMudrak, B., & Kuehn, M. J. (2010). Heat-Labile Enterotoxin: Beyond G M1 Binding. Toxins, 2(6), 1445-1470. https://doi.org/10.3390/toxins2061445