Structure-Based Design of Ricin Inhibitors

Abstract
:1. Introduction
2. Ricin Structure and Action: Implications for Inhibitor Design
2.1. X-Ray Structure of Ricin
2.2. RTB Is Not a Good Prospect for Structure-Based Inhibitor Design
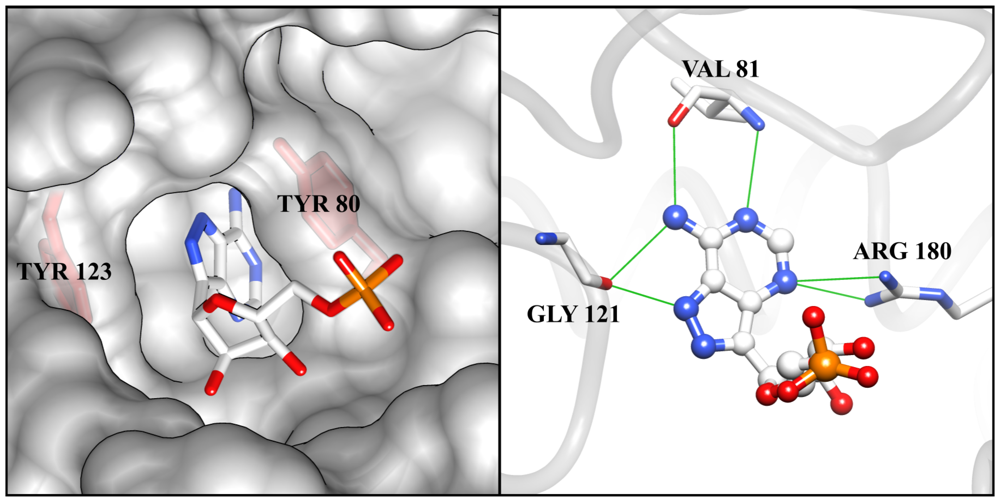
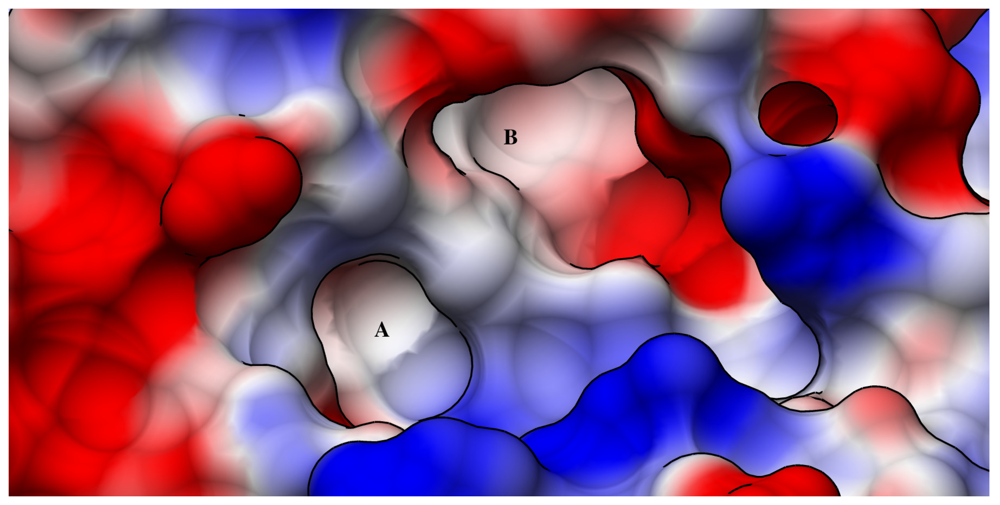
2.3. RTA Is a Plausible, but Challenging Inhibitor Design Target


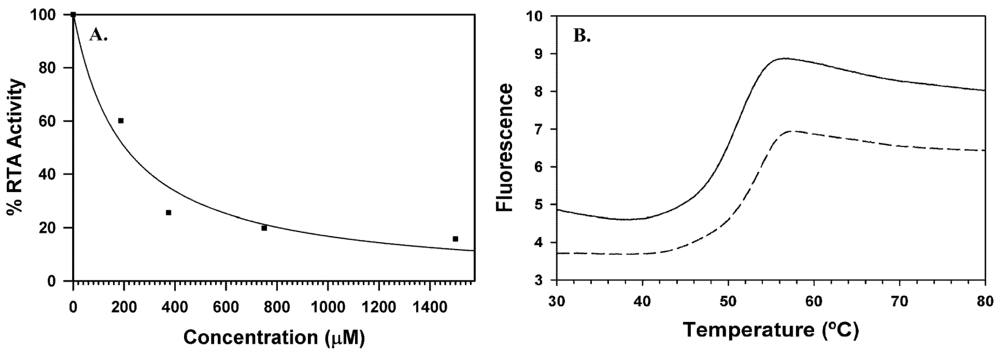
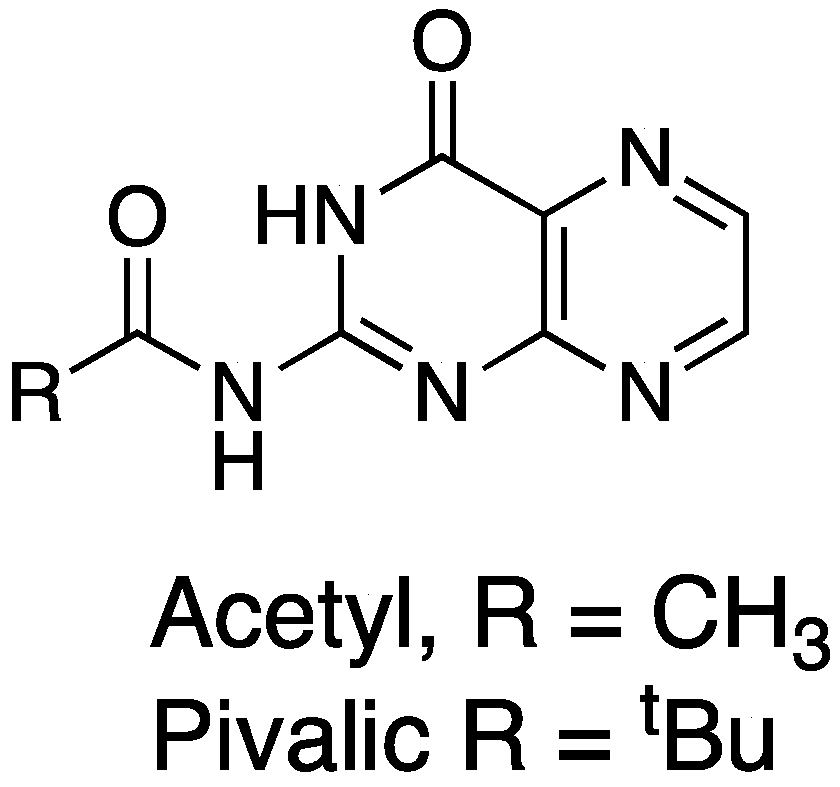
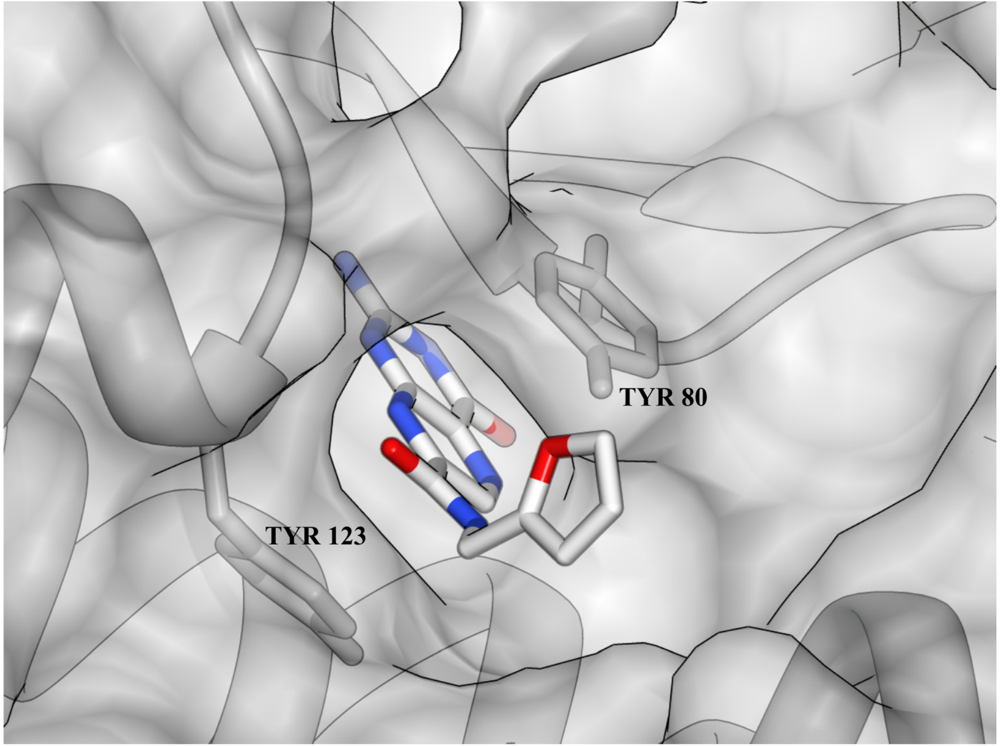
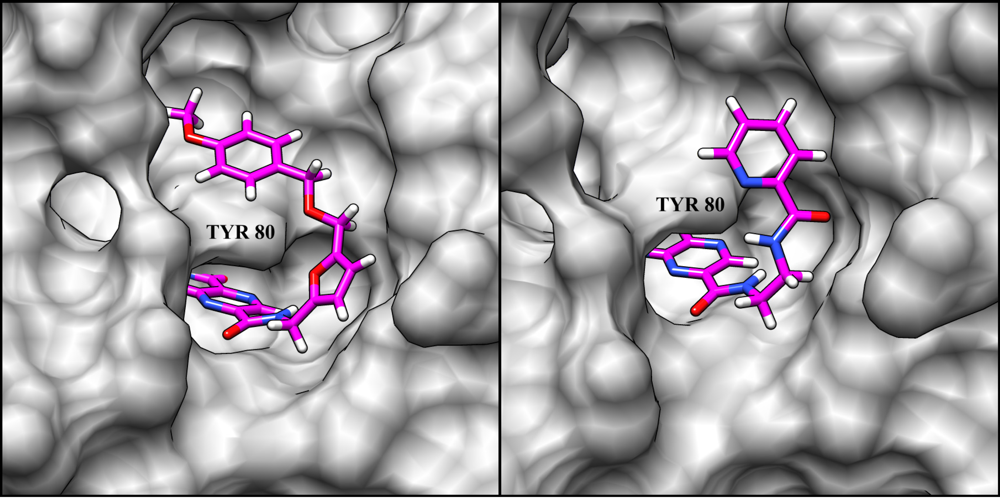
3. Ricin Inhibitors


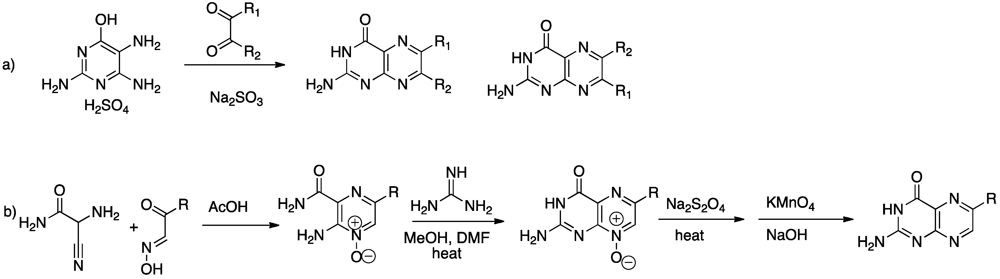
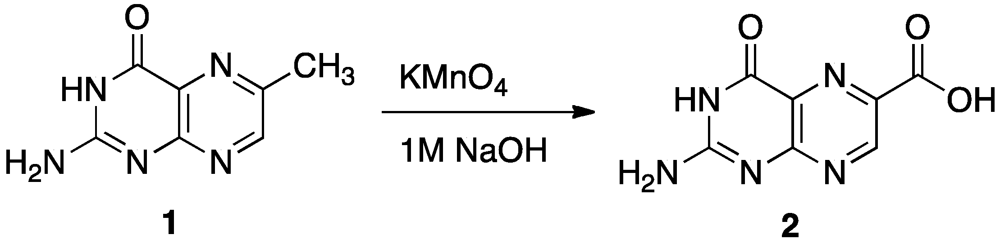
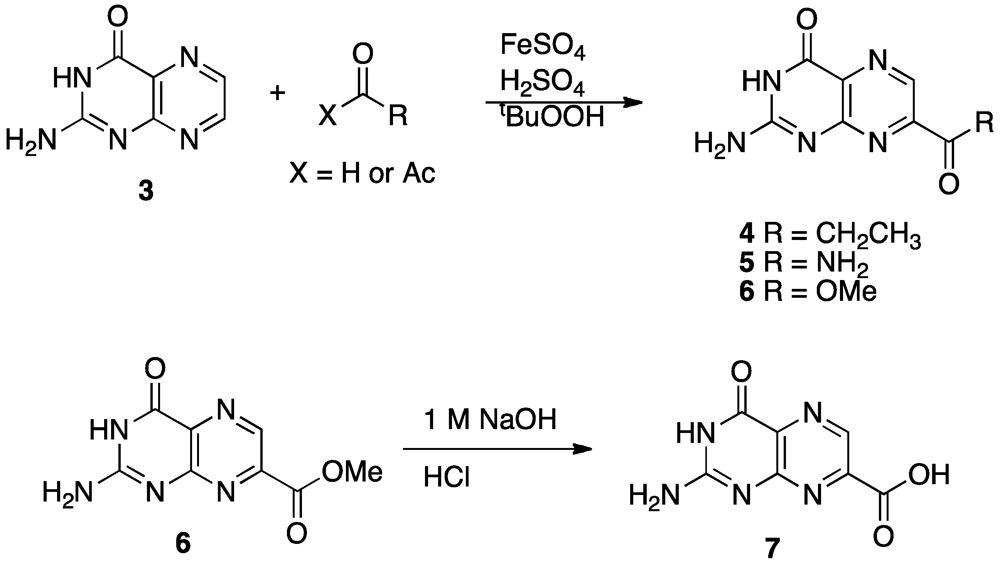
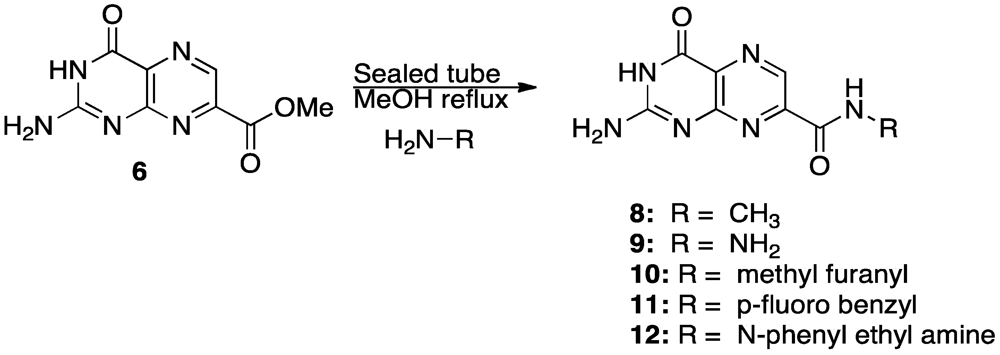
4. Novel Compound Synthesis






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Structure | Name | IC50 * | Resolution |
|---|---|---|---|---|
| PTA |  | Pteroic acid | 600 μM | 2.30 Å |
| 1 |  | 6-methyl pterin | No Inhibition | NA |
| 2 |  | 6-carboxy pterin | No Inhibition | NA |
| 7 |  | 7-carboxy pterin (7CP) | 240 μM | 1.29 Å |
| 5 |  | 7-carbamoyl pterin | No Inhibition | 1.75 Å |
| 8 |  | N-methyl-7-carbamoyl pterin | 1.6 mM | 1.26 Å |
| 9 |  | 7-hydrazide pterin | 500 μM (35%) | 1.97 Å |
| 4 |  | 7-propionyl pterin | 750 μM | 1.35 Å |
| 10 |  | N-(furanylmethyl)-7-carbamoyl pterin | 380 μM | 1.89 Å |
| 11 |  | N-(4-fluorobenzyl)-7-carbamoyl pterin | 570 μM | NA |
| 12 |  | N-(2-(phenylamino) ethyl)-7-carbamoyl pterin | 200 μM | 1.75 Å |
5. Plans for Inhibitor Optimization


Acknowledgements
References
- Olsnes, S.; Pihl, A. Toxic Lectins and Related Proteins. In The Molecular Action of Toxins and Viruses; Coen, P., van Heynigen, S., Eds.; Elsevier Biomedical Press: New York, NY, USA, 1982. [Google Scholar]
- Stirpe, F.; Barbieri, L. Ribosome-inactivating proteins up to date. FEBS Lett. 1986, 195, 1–8. [Google Scholar]
- Ready, M.P.; Brown, D.T.; Robertus, J.D. Extracellular localization of pokeweed antiviral protein. Proc. Natl. Acad. Sci. USA 1986, 83, 5053–5056. [Google Scholar]
- Burnham, R.M. Threat of Bioterrorism in America. In Subcommittee on Oversight and Investigations; Report before the U.S. House of Representatives, 1999. Available online: http://www.fas.org/irp/congress/1999_hr/990520-bioleg3.htm (accessed on 30 September 2011).
- Rich, V. Murderous experiments of stalin’s police chief. New Sci. 1992, 135, 8. [Google Scholar]
- Mishra, V.; Bilgrami, S.; Sharma, R.S.; Kaur, P.; Yadav, S.; Krauspenhaar, R.; Betzel, C.; Voelter, W.; Babu, C.R.; Singh, T.P. Crystal structure of himalayan mistletoe ribosome-inactivating protein reveals the presence of a natural inhibitor and a new functionally active sugar-binding site. J. Biol. Chem. 2005, 280, 20712–20721. [Google Scholar]
- Montfort, W.; Villafranca, J.E.; Monzingo, A.F.; Ernst, S.R.; Katzin, B.; Rutenber, E.; Xuong, N.H.; Hamlin, R.; Robertus, J.D. The three-dimensional structure of ricin at 2.8 A. J. Biol. Chem. 1987, 262, 5398–5403. [Google Scholar] [PubMed]
- Rutenber, E.; Katzin, B.J.; Ernst, S.; Collins, E.J.; Mlsna, D.; Ready, M.P.; Robertus, J.D. Crystallographic refinement of ricin to 2.5 A. Proteins 1991, 10, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Monzingo, A.F.; Robertus, J.D. X-ray analysis of substrate analogs in the ricin A-chain active site. J. Mol. Biol. 1992, 227, 1136–1145. [Google Scholar]
- Ho, M.C.; Sturm, M.B.; Almo, S.C.; Schramm, V.L. Transition state analogues in structures of ricin and saporin ribosome-inactivating proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 20276–20281. [Google Scholar]
- Katzin, B.J.; Collins, E.J.; Robertus, J.D. Structure of ricin A-chain at 2.5 A. Proteins 1991, 10, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Olsnes, S.; Fernandez-Puentes, C.; Carrasco, L.; Vazquez, D. Ribosome inactivation by the toxic lectins abrin and ricin. Kinetics of the enzymic activity of the toxin A-chains. Eur. J. Biochem. 1975, 60, 281–288. [Google Scholar] [CrossRef]
- Weston, S.A.; Tucker, A.D.; Thatcher, D.R.; Derbyshire, D.J.; Pauptit, R.A. X-ray structure of recombinant ricin A-chain at 1.8 A resolution. J. Mol. Biol. 1994, 244, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Robertus, J.D.; Monzingo, A.F. The structure of ribosome inactivating proteins. Mini Rev. Med. Chem. 2004, 4, 477–486. [Google Scholar]
- Mlsna, D.; Monzingo, A.F.; Katzin, B.J.; Ernst, S.; Robertus, J.D. Structure of recombinant ricin A chain at 2.3 A. Protein Sci. 1993, 2, 429–435. [Google Scholar] [PubMed]
- Hajduk, P.J.; Huth, J.R.; Fesik, S.W. Druggability indices for protein targets derived from NMR-based screening data. J. Med. Chem. 2005, 48, 2518–2525. [Google Scholar]
- Wahome, P.G.; Bai, Y.; Neal, L.M.; Robertus, J.D.; Mantis, N.J. Identification of small-molecule inhibitors of ricin and shiga toxin using a cell-based high-throughput screen. Toxicon 2010, 56, 313–323. [Google Scholar]
- Sandvig, K.; Olsnes, S.; Pihl, A. Kinetics of binding of the toxic lectins abrin and ricin to surface receptors of human cells. J. Biol. Chem. 1976, 251, 3977–3984. [Google Scholar]
- Laurie, A.T.; Jackson, R.M. Q-SiteFinder: an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics 2005, 21, 1908–1916. [Google Scholar]
- Endo, Y.; Tsurugi, K. RNA N-glycosidase activity of ricin A-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J. Biol. Chem. 1987, 262, 8128–8130. [Google Scholar] [PubMed]
- Endo, Y.; Tsurugi, K. The RNA N-glycosidase activity of ricin A-chain. The characteristics of the enzymatic activity of ricin A-chain with ribosomes and with rRNA. J. Biol. Chem. 1988, 263, 8735–8739. [Google Scholar] [PubMed]
- Ready, M.P.; Kim, Y.; Robertus, J.D. Site-directed mutagenesis of ricin A-chain and implications for the mechanism of action. Proteins 1991, 10, 270–278. [Google Scholar]
- Chen, X.Y.; Link, T.M.; Schramm, V.L. Ricin A-chain: kinetics, mechanism, and RNA stem-loop inhibitor. Biochemistry 1998, 37, 11605–11613. [Google Scholar]
- Cheng, A.C.; Coleman, R.G.; Smyth, K.T.; Cao, Q.; Soulard, P.; Caffrey, D.R.; Salzberg, A.C.; Huang, E.S. Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 2007, 25, 71–75. [Google Scholar]
- Wahome, P.G.; Bai, Y.; Neal, L.M.; Robertus, J.D.; Mantis, N.J. Identification of small-molecule inhibitors of ricin and shiga toxin using a cell-based high-throughput screen. Toxicon 2010, 56, 313–323. [Google Scholar]
- Lord, J.M.; Deeks, E.; Marsden, C.J.; Moore, K.; Pateman, C.; Smith, D.C.; Spooner, R.A.; Watson, P.; Roberts, L.M. Retrograde transport of toxins across the endoplasmic reticulum membrane. Biochem. Soc. Trans. 2003, 31, 1260–1262. [Google Scholar]
- Stechmann, B.; Bai, S.K.; Gobbo, E.; Lopez, R.; Merer, G.; Pinchard, S.; Panigai, L.; Tenza, D.; Raposo, G.; Beaumelle, B.; Sauvaire, D.; Gillet, D.; Johannes, L.; Barbier, J. Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell 2010, 141, 231–242. [Google Scholar]
- Roday, S.; Amukele, T.; Evans, G.B.; Tyler, P.C.; Furneaux, R.H.; Schramm, V.L. Inhibition of ricin A-chain with pyrrolidine mimics of the oxacarbenium ion transition state. Biochemistry 2004, 43, 4923–4933. [Google Scholar]
- Bai, Y.; Monzingo, A.F.; Robertus, J.D. The X-ray structure of ricin A chain with a novel inhibitor. Arch. Biochem. Biophys. 2009, 483, 23–28. [Google Scholar]
- Miller, D.J.; Ravikumar, K.; Shen, H.; Suh, J.K.; Kerwin, S.M.; Robertus, J.D. Structure-based design and characterization of novel platforms for ricin and shiga toxin inhibition. J. Med. Chem. 2002, 45, 90–98. [Google Scholar]
- Yan, X.; Hollis, T.; Svinth, M.; Day, P.; Monzingo, A.F.; Milne, G.W.; Robertus, J.D. Structure-based identification of a ricin inhibitor. J. Mol. Biol. 1997, 266, 1043–1049. [Google Scholar]
- Chang, C.E.; Chen, W.; Gilson, M.K. Ligand configurational entropy and protein binding. Proc. Natl. Acad. Sci. USA 2007, 104, 1534–1539. [Google Scholar]
- Vedadi, M.; Niesen, F.H.; Allali-Hassani, A.; Fedorov, O.Y.; Finerty, P.J., Jr.; Wasney, G.A.; Yeung, R.; Arrowsmith, C.; Ball, L.J.; Berglund, H.; et al. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. USA 2006, 103, 15835–15840. [Google Scholar]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar]
- Chen, X.-Y.; Berti, P.J.; Schramm, V.L. Transition-State Analysis for Depurination of DNA by Ricin A-Chain. J. Am. Chem. Soc. 2000, 122, 6527–6534. [Google Scholar]
- Waring, P.; Armarego, W.L.F. 10. A New Preparation of 6-Hydroxymethylpterin from 6-Methylpterin. Aust. J. Chem. 1985, 38, 629–631. [Google Scholar]
- Taylor, E.C.; Kobylecki, R. Pteridines. 43. Facile Synthesis of 6-Chloropterin and 2,4-Diamino-6-Chloropteridine. J. Org. Chem. 1978, 43, 680–683. [Google Scholar]
- Taylor, E.C.; Jacobi, P.A. Letter: An unequivocal total synthesis of L-erythro-biopterin. J. Am. Chem. Soc. 1974, 96, 6781–6782. [Google Scholar]
- Taylor, E.C.; Henrie, R.N.; Portnoy, R.C. Pteridines .44. Convenient Synthesis of 6-Formylpterin. J. Org. Chem. 1978, 43, 736–737. [Google Scholar]
- Storm, C.B.; Shiman, R.; Kaufman, S. Preparation of 6-Substituted Pterins via Isay Reaction. J. Org. Chem. 1971, 36, 3925. [Google Scholar]
- Taylor, E.C.; Jacobi, P.A. Pteridines. XXX. A facile synthesis of xanthopterin. J. Am. Chem. Soc. 1973, 95, 4455–4456. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.C.; Otiv, S.R.; Durucasu, I. Protection and Deprotection of Fused 2-Amino-4(3h)-Pyrimidinones-Conversion of Pterins and 5-Deazapterins to 2,4-Diamino Derivatives. Heterocycles 1993, 36, 1883–1895. [Google Scholar]
- Pfleider, W.; Zondler, H.; Mengel, R. Pteridines .39. Synthesis and Structure of Pterin-6-Carboxylic and Pterin-7-Carboxylic Acids. Annalen Der Chemie-Justus Liebig 1970, 741, 64. [Google Scholar]
- Caronna, T.; Gardini, G.P.; Minisci, F. Nucleophilic Character of Acyl Radicals-a New Selective Type of Aromatic Acylation. J. Chem. Soc. D-Chem. Commun. 1969, 5, 201. [Google Scholar]
- Caronna, T.; Minisci, F.; Porta, O.; Fronza, G. Homolytic Acylation of Protonated Pyridine and Pyrazine Derivatives. J. Chem. Soc.-Perkin Trans. 2 1972, 14, 2035. [Google Scholar]
- Pruet, J.M.; Robertus, J.D.; Anslyn, E.V. Acyl radical insertion for the direct formation of new 7-substituted pterin analogs. Tetrahedron Lett. 2010, 51, 2539–2540. [Google Scholar]
- Houminer, Y.; Southwick, E.W.; Williams, D.L. Substituent-Directing Effects in the Homolytic Acylation of Pyrazine Derivatives. J. Org. Chem. 1989, 54, 640–643. [Google Scholar]
- Pruet, J.M.; Jasheway, K.R.; Manzano, L.A.; Bai, Y.; Anslyn, E.V.; Robertus, J.D. 7-Substituted pterins provide a new direction for ricin A chain inhibitors. Eur. J. Med. Chem 2011, in press. [Google Scholar]
- Warren, G.L.; Andrews, C.W.; Capelli, A.M.; Clarke, B.; Lalonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; Tedesco, G.; Wall, I.D.; Woolven, J.M.; Peishoff, C.E.; Head, M.S. A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar]
- Kellenberger, E.; Rodrigo, J.; Muller, P.; Rognan, D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins 2004, 57, 225–242. [Google Scholar]
- Kontoyianni, M.; McClellan, L.M.; Sokol, G.S. Evaluation of docking performance: comparative data on docking algorithms. J. Med. Chem. 2004, 47, 558–565. [Google Scholar]
- Cournia, Z.; Leng, L.; Gandavadi, S.; Du, X.; Bucala, R.; Jorgensen, W.L. Discovery of human macrophage migration inhibitory factor (MIF)-CD74 antagonists via virtual screening. J. Med. Chem. 2009, 52, 416–424. [Google Scholar]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar]
- Abagyan, R.A.; Totrov, M.; Kuznetsov, D. ICM-a new method for protein modeling and design: Applications to docking and structure prediction from the disordered native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jasheway, K.; Pruet, J.; Anslyn, E.V.; Robertus, J.D. Structure-Based Design of Ricin Inhibitors. Toxins 2011, 3, 1233-1248. https://doi.org/10.3390/toxins3101233
Jasheway K, Pruet J, Anslyn EV, Robertus JD. Structure-Based Design of Ricin Inhibitors. Toxins. 2011; 3(10):1233-1248. https://doi.org/10.3390/toxins3101233
Chicago/Turabian StyleJasheway, Karl, Jeffrey Pruet, Eric V. Anslyn, and Jon D. Robertus. 2011. "Structure-Based Design of Ricin Inhibitors" Toxins 3, no. 10: 1233-1248. https://doi.org/10.3390/toxins3101233
APA StyleJasheway, K., Pruet, J., Anslyn, E. V., & Robertus, J. D. (2011). Structure-Based Design of Ricin Inhibitors. Toxins, 3(10), 1233-1248. https://doi.org/10.3390/toxins3101233