Synthesis and Analgesic Effects of μ-TRTX-Hhn1b on Models of Inflammatory and Neuropathic Pain


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
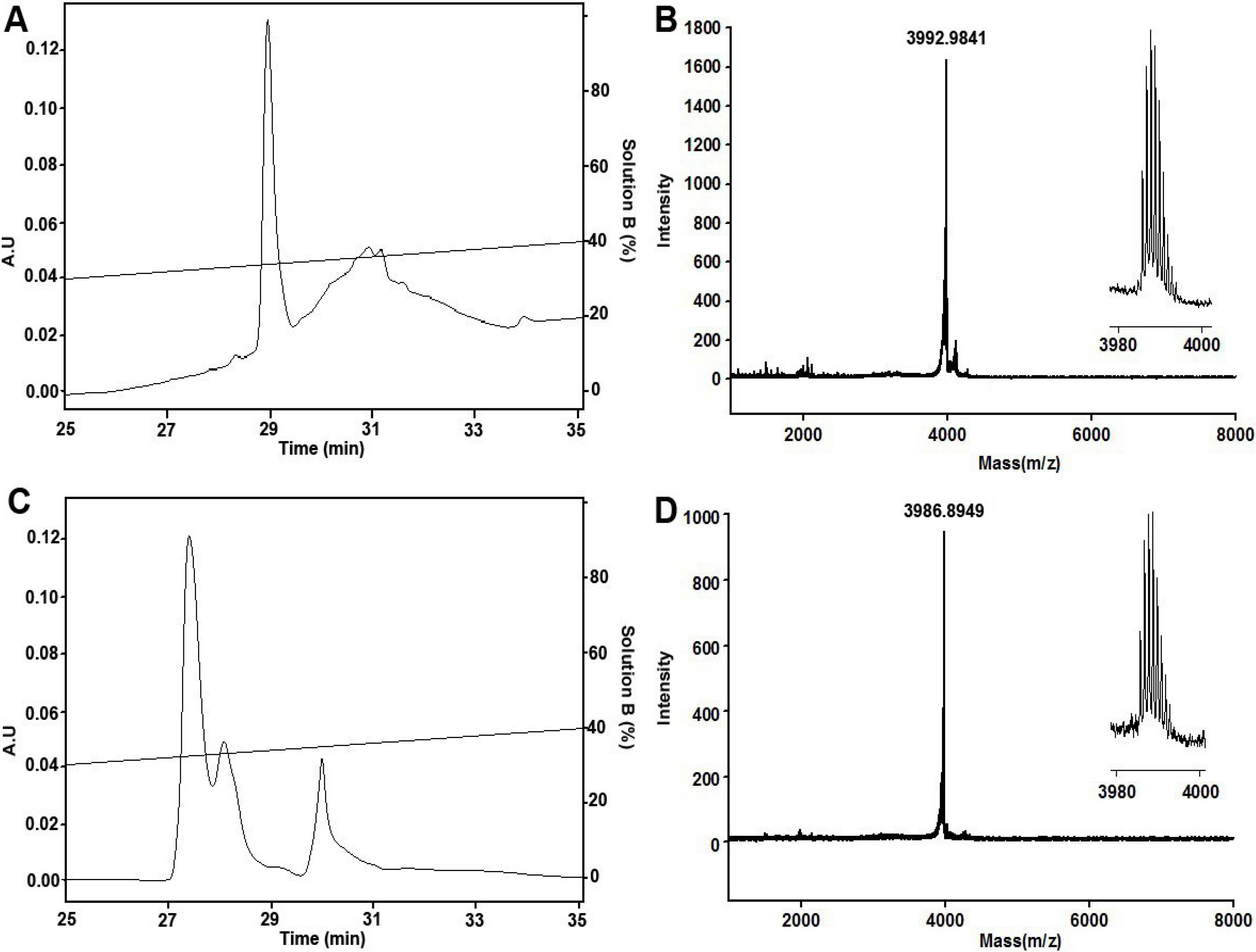
2.1. Peptide Synthesis, Folding and Characterization

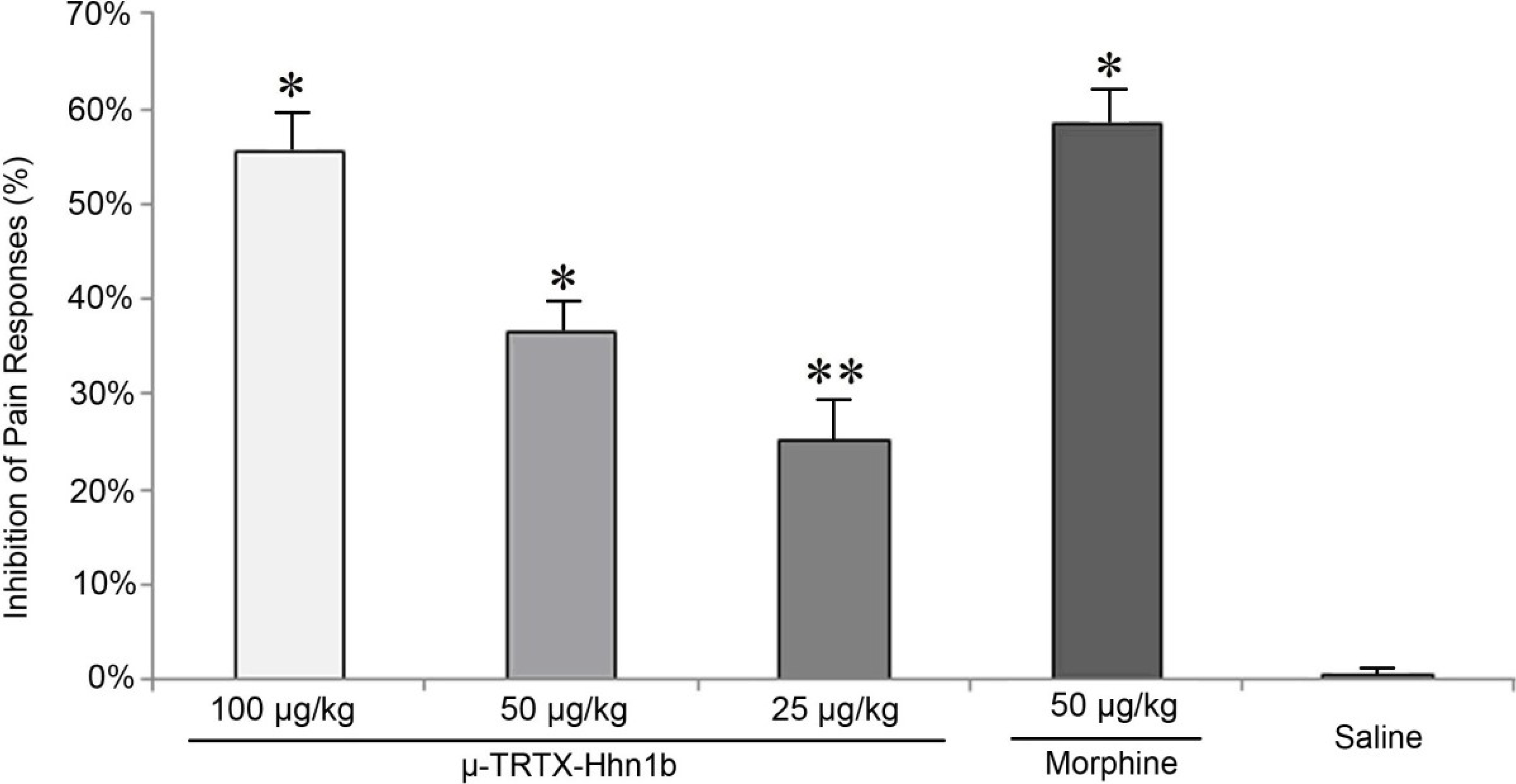
2.2. The Analgesic Effect of μ-TRTX-Hhn1b on the Acetic Acid-Induced Abdominal Constriction Mouse Model


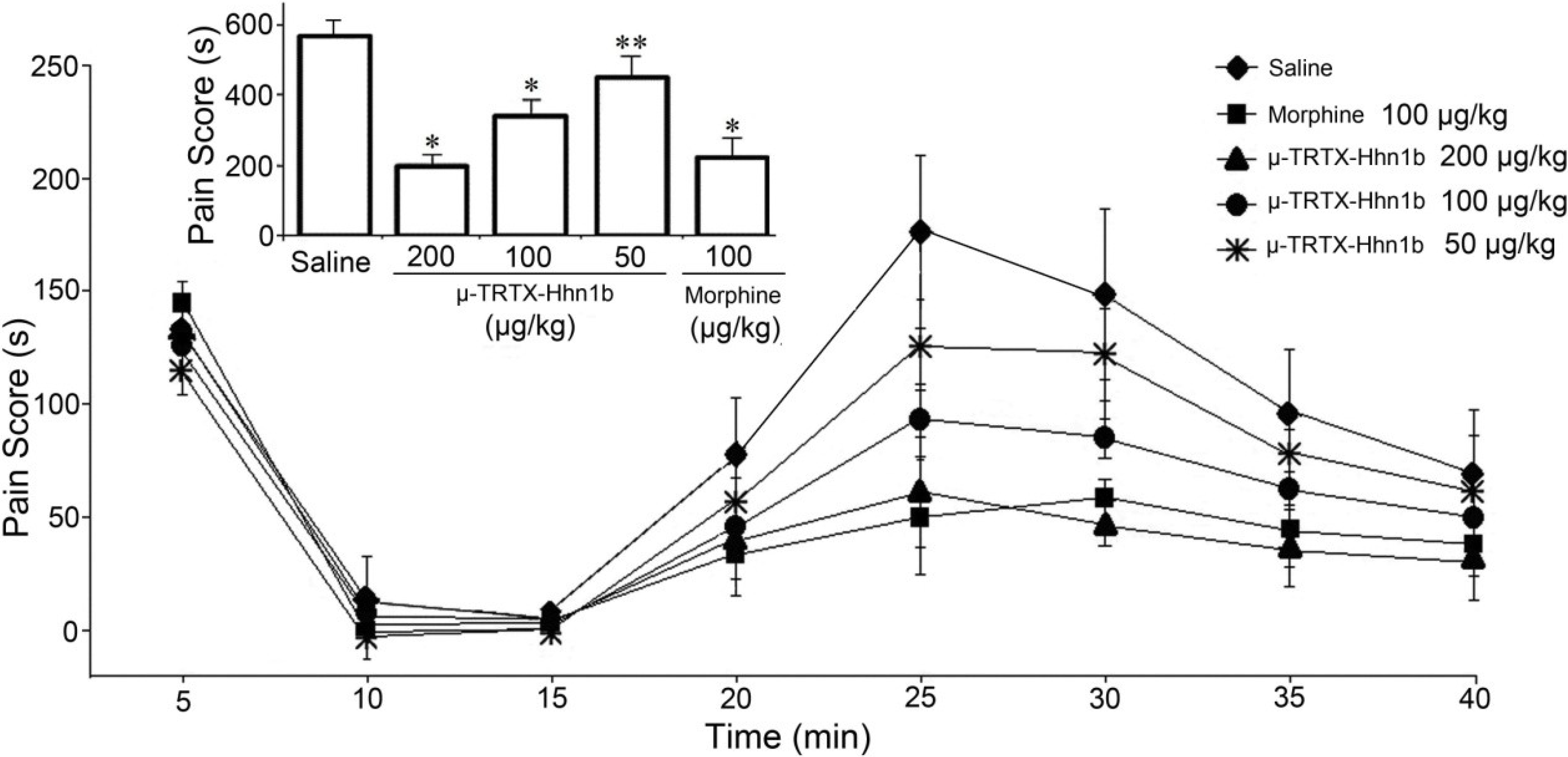
2.3. The Analgesic Effect of μ-TRTX-Hhn1b on the Formalin-Induced Inflammatory Rat Model

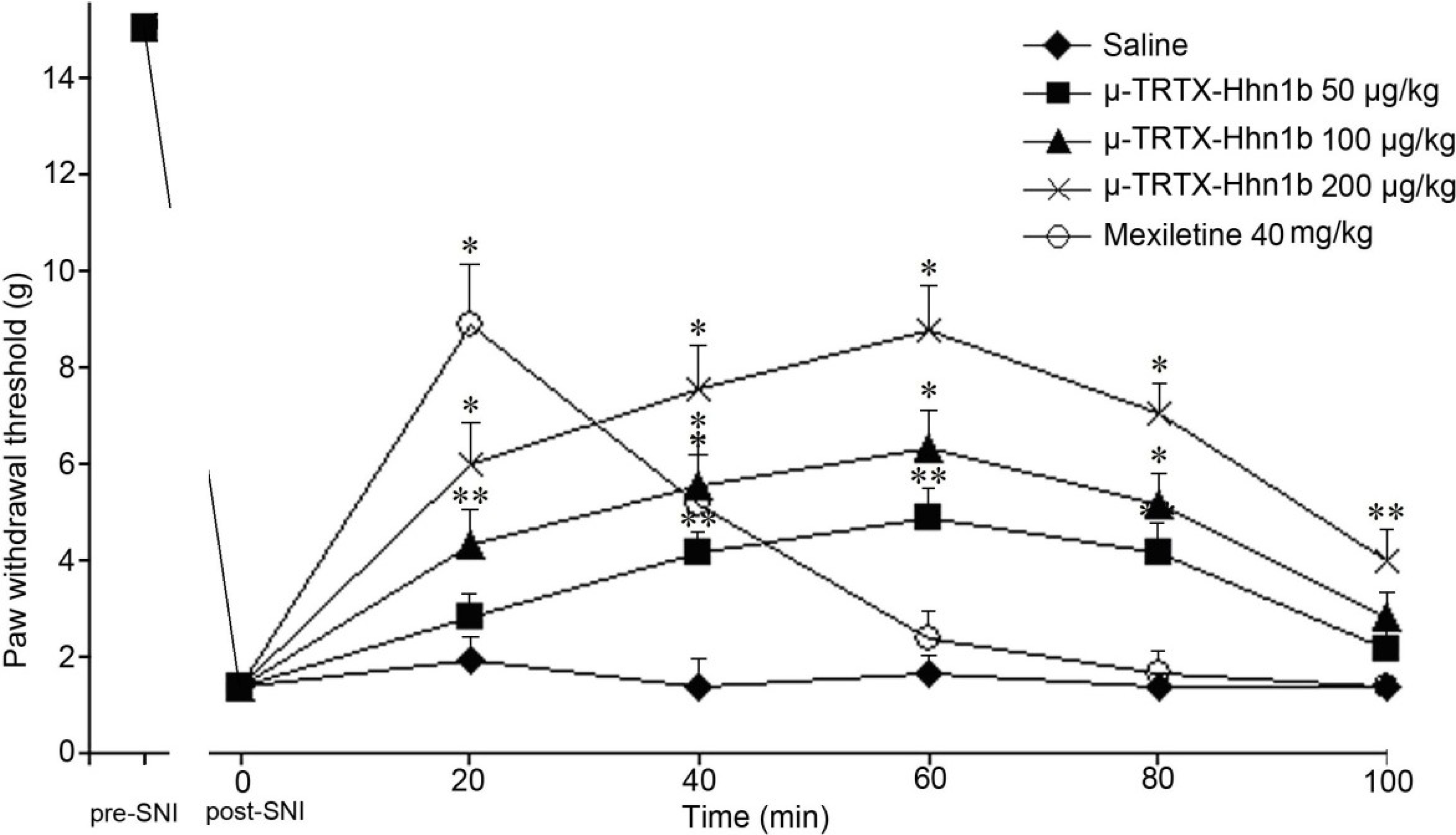
2.4. The Analgesic Effect of μ-TRTX-Hhn1b on Spinal Nerve Injury Rat Model

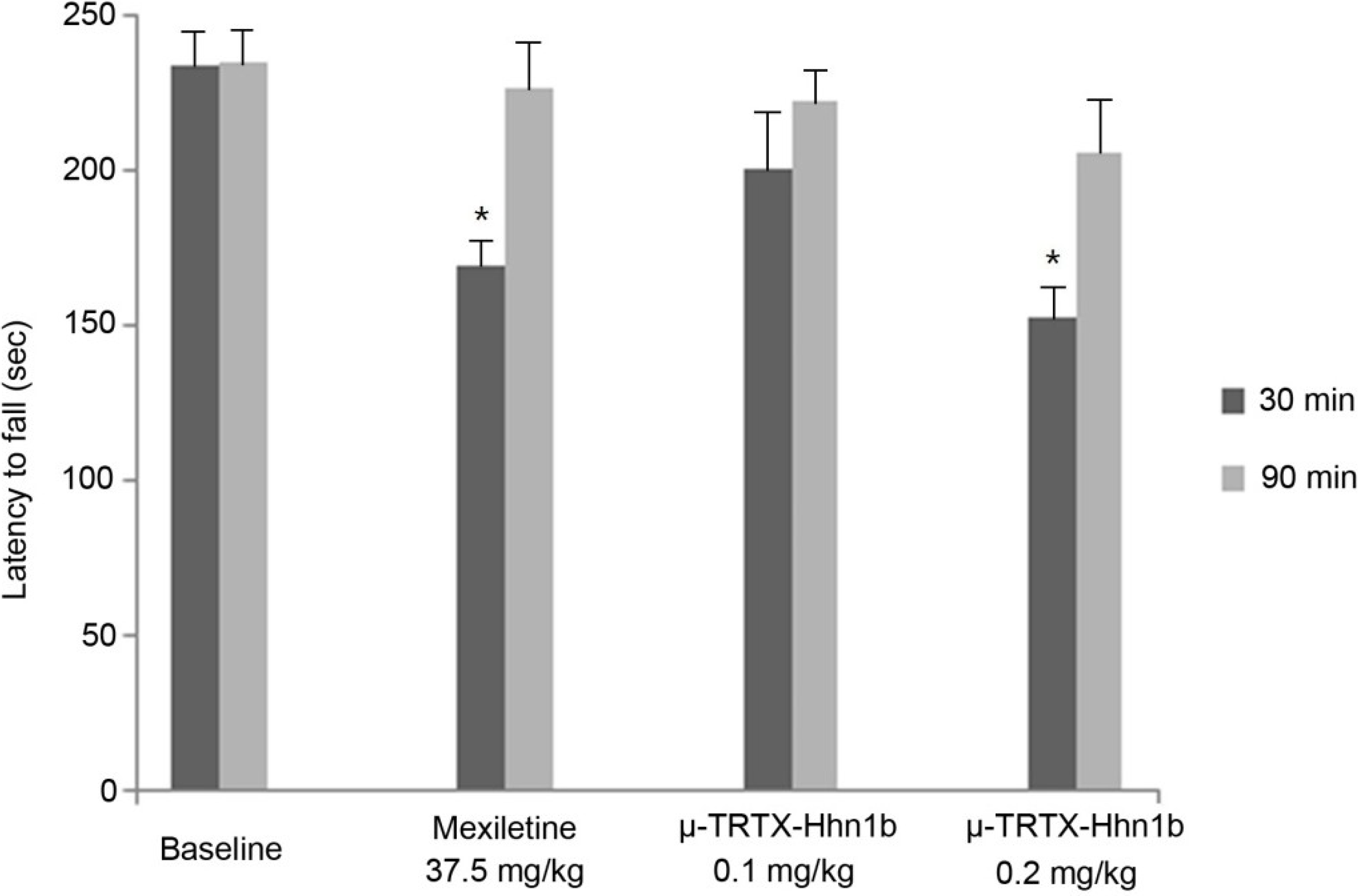
2.5. Rat Rotarod Model of Motor Coordination

3. Experimental Section
3.1. Peptide Synthesis, Folding and Purification
3.2. Electrophysiological Assays
3.3. Animal
3.4. Abdominal Constriction Response Caused by Acetic Acid
3.5. Formalin Test
3.6. Spinal Nerve Injury Model
3.7. Rotarod Model of Motor Coordination
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Catterall, W.A. Voltage-gated sodium channels at 60: Structure, function, and pathophysiology. J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [CrossRef]
- Cummins, T.R.; Rush, A.M. Voltage-gated sodium channel blockers for the treatment of neuropathic pain. Expert. Rev. Neurother. 2007, 7, 1597–1612. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 2010, 33, 325–347. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Black, J.A.; Waxman, S.G. Voltage-gated sodium channels: Therapeutic targets for pain. Pain Med. 2009, 10, 1260–1269. [Google Scholar] [CrossRef]
- Wang, J.; Yarov-Yarovoy, V.; Kahn, R.; Gordon, D.; Gurevitz, M.; Scheuer, T.; Gatterall, W.A. Mapping the receptor site for alpha-scorpion toxins on a Na+ channel voltage sensor. Proc. Natl. Acad. Sci. USA 2011, 108, 15426–15431. [Google Scholar]
- Zhang, J.Z.; Yarov-Yarovoy, V.; Scheuer, T.; Karbat, I.; Cohen, L.; Gordon, D.; Gurevitz, M.; Catterall, W.A. Structure-function map of the receptor site for b-scorpion toxins in domain II of voltage-gated sodium channels. J. Biol. Chem. 2011, 286, 33641–33651. [Google Scholar] [CrossRef]
- Liu, Y.; Li, D.; Wu, Z.; Li, J.; Nie, D.S.; Xiang, Y.; Liu, Z.H. A positively charged surface patch is important for hainantoxin-IV binding to voltage-gated sodium channels. J. Pept. Sci. 2012, 18, 643–649. [Google Scholar] [CrossRef]
- Drenth, J.P.; Waxman, S.G. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J. Clin. Investig. 2007, 117, 3603–3609. [Google Scholar] [CrossRef]
- Fertleman, C.R.; Baker, M.D.; Parker, K.A.; Moffatt, S.; Elmslie, F.V.; Abrahamsen, B.; Ostman, J.; Klugbauer, N.; Wood, J.N.; Gardiner, R.M.; et al. SCN9A mutations in paroxysmal extreme pain disorder: Allelic variants underlie distinct channel defects and phenotypes. Neuron 2006, 52, 767–774. [Google Scholar] [CrossRef]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Estacion, M.; Jarecki, B.W.; Tyrrell, L.; Fischer, T.Z.; Lawden, M.; Cummins, T.R.; Waxman, S.G. Paroxysmal extreme pain disorder M1627K mutation in human Nav1.7 renders DRG neurons hyperexcitable. Mol. Pain 2008, 4. [Google Scholar] [CrossRef]
- Estacion, M.; Dib-Hajj, S.D.; Benke, P.J.; Te Morsche, R.H.; Eastman, E.M.; Macala, L.J.; Drenth, J.P.; Waxman, S.G. Nav1.7 gain-of-function mutations as a continuum: A1632E displays physiological changes associated with erythromelalgia and paroxysmal extreme pain disorder mutations and produces symptoms of both disorders. J. Neurosci. 2008, 28, 11079–11088. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. From genes to pain: Nav1.7 and human pain disorders. Trends Neurosci. 2007, 30, 555–563. [Google Scholar] [CrossRef]
- Sheets, P.L.; Jackson, J.O.; Waxman, S.G.; Dib-Hajj, S.; Cummins, T.R. A Nav1.7 channel mutation associated with hereditary erythromelalgia contributes to neuronal hyperexcitability and displays reduced lidocaine sensitivity. J. Physiol. 2007, 581, 1019–1031. [Google Scholar] [CrossRef]
- Ahmad, S.; Dahllund, L.; Eriksson, A.B.; Hellgren, D.; Karlsson, U.; Lund, P.E.; Meijer, I.A.; Meury, L.; Mills, T.; Moody, A.; et al. A stop codon mutation in SCN9A causes lack of pain sensation. Hum. Mol. Genet. 2007, 16, 2114–2121. [Google Scholar] [CrossRef]
- Nilsen, K.B.; Nicholas, A.K.; Woods, C.G.; Mellgren, S.I.; Nebuchennykh, M.; Aasly, J. Two novel SCN9A mutations causing insensitivity to pain. Pain 2009, 143, 155–158. [Google Scholar]
- Cestele, S.; Catterall, W.A. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 2000, 82, 883–892. [Google Scholar] [CrossRef]
- Bosmans, F.; Swartz, K.J. Targeting voltage sensors in sodium channels with spider toxins. Trends Pharmacol. Sci. 2010, 31, 175–182. [Google Scholar] [CrossRef]
- Saez, N.J.; Senff, S.; Jensen, J.E.; Er, S.Y.; Herzig, V.; Rash, L.D.; King, G.F. Spider-venom peptides as therapeutics. Toxins 2010, 12, 2851–2871. [Google Scholar]
- Billen, B.; Bosmans, F.; Tytgat, J. Animal peptides targeting voltage activated sodium channels. Curr. Pharm. Des. 2008, 14, 2492–2502. [Google Scholar] [CrossRef]
- Catterall, W.A.; Cestèle, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [Green Version]
- Klint, J.K.; Senff, S.; Rupasinghe, D.B.; Er, S.Y.; Herzig, V.; Nicholson, G.M.; King, G.F. Spider-venom peptides that target voltage-gated sodium channels: Pharmacological tools and potential therapeutic leads. Toxicon 2012, 60, 478–491. [Google Scholar] [CrossRef]
- Wood, D.L.; Miljenović, T.; Cai, S.; Raven, R.J.; Kaas, Q.; Escoubas, P.; Herzig, V.; Wilson, D.; King, G.F. ArachnoServer: A database of protein toxins from spiders. BMC Genomics 2009, 10. [Google Scholar] [CrossRef]
- Norman, I.P. The World Spider Catalog; Version 15; American Museum of Natural History: New York, NY, USA, 2014. [Google Scholar]
- Liu, Z.H.; Dai, J.; Chen, Z.R.; Hu, W.J.; Xiao, Y.C.; Liang, S.P. Isolation and characterization of hainantoxin-IV, a novel antagonist tetrodotoxinsensitive sodium channels form the Chinese bird spider Ornithoctorus hainana. Cell. Mol. Life Sci. 2003, 60, 972–978. [Google Scholar] [CrossRef]
- Li, D.L.; Xiao, Y.C.; Xu, X.; Xiong, X.; Lu, S.; Liu, Z.; Zhu, Q.; Wang, M.; Gu, X.; Liang, S.; et al. Structure-activity relationships of hainantoxin-IV and structure determination of active and inactive sodium channel blockers. J. Biol. Chem. 2004, 279, 37734–37740. [Google Scholar] [CrossRef]
- Liu, Z.H.; Hunan Normal University, Changsha, China. Unpublished work. 2014.
- Collier, H.O.J.; Dinneen, J.C.; Johnson, C.A.; Schneider, C. The abdominal constriction response and its suppression by analgesic drugs in the mouse. Br. J. Pharmacol. Chemother. 1968, 32, 295–310. [Google Scholar] [CrossRef]
- Vinegar, R.; Truax, J.F.; Selph, J.L.; Johnston, P.R. Antagonism of pain and hyperalgesia. Anti-inflammatory Drugs. In Handbook of Experimental Pharmacology; Vane, J.R., Ferreira, S.H., Eds.; Springer-Verlag: Berlin, Germany, 1979; Volume 50/II, pp. 208–222. [Google Scholar]
- Kumazawa, T.; Mizumura, K.; Koda, H.; Fukusako, H. EP receptor subtypes implicated in the PGE2-induced sensitization of polymodal receptors in response to bradykinin and heat. J. Neurophysiol. 1996, 75, 2361–2368. [Google Scholar]
- Correa, C.R.; Kyle, D.J.; Chakraverty, S.; Calixto, J.B. Antinociceptive profile of the pseudopeptide B2 bradykinin receptor antagonist NPC 18688 in mice. Br. J. Pharmacol. 1996, 117, 552–558. [Google Scholar]
- Goettl, V.M.; Larson, A.A. An antagonist of substance P N-terminal fragments, D-substance P(1–7), reveals that both nociceptive and antinociceptive effects are induced by substance P N-terminal activity during noxious chemical stimulation. Brain Res. 1998, 780, 80–85. [Google Scholar]
- Ferrira, S.H.; Vane, S.R. New aspects on the model of action of nonsteroid anti-inflammatory drugs. Annu. Rev. Pharmacol. 2005, 98, 267–273. [Google Scholar]
- Perianayagam, J.B.; Sharma, S.K.; Joseph, A.; Christina, A.J. Evaluation of antipyretic and analgesic activity of Emblica officinalis Gaertn. J. Ethnopharmacol. 2004, 95, 83–85. [Google Scholar] [CrossRef]
- Hong, Y.; Abbott, F.V. Peripheral opioid modulation of pain and inflammation in the formalin test. Eur. J. Pharmacol. 1995, 277, 21–28. [Google Scholar] [CrossRef]
- Moncada, S.; Ferreira, S.H.; Vane, J.R. Inhibition of prostaglandin biosynthesis as the mechanism of analgesia of aspirin-like drugs in the dog knee joint. Eur. J. Pharmacol. 1975, 31, 250–260. [Google Scholar] [CrossRef]
- Bennett, G.J.; Xie, Y.K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [Google Scholar] [CrossRef]
- Decosterd, I.; Woolf, C.J. Spared nerve injury: An animal model of persistent peripheral neuropathic pain. Pain 2000, 87, 149–158. [Google Scholar] [CrossRef]
- Erichsen, H.K.; Hao, J.X.; Xu, X.J.; Blackburn-Munro, G. A comparison of the antinociceptive effects of voltage-activated Na+ channel blockers in two rat models of neuropathic pain. Eur. J. Pharmacol. 2003, 458, 275–282. [Google Scholar] [CrossRef]
- Dunham, N.W.; Miya, T.S. A note on a simple apparatus for detecting neurological deficit in rats and mice. J. Am. Pharm. Assoc. 1957, 46, 208–209. [Google Scholar] [CrossRef]
- Goldin, A.L. Resurgence of sodium channel research. Ann. Rev. Physiol. 2011, 63, 871–894. [Google Scholar] [CrossRef]
- Burgess, D.L.; Kohrman, D.C.; Galt, J.; Plummer, N.W.; Jones, J.M. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant “motor endplate disease”. Nat. Genet. 1995, 10, 461–465. [Google Scholar] [CrossRef]
- Ruth, S.; Angelika, L.; Tobias, H.; Andrea, S.L.; Johannes, F.; Christian, A.; Peter, G.; Richard, W.C. Anticancer drug oxaliplatin induces acute cooling-aggravated neuropathy via sodium channel subtype Nav1.6-regurgent and persistent current. Proc. Natl. Acad. Sci. 2012, 109, 6704–6709. [Google Scholar] [CrossRef]
- Zhu, Q.; Liang, S.P.; Martin, L.; Gasparini, S.; Mènez, A.; Vita, C. Role of disulfide bonds in folding and activity of leiurotoxin I: Just two disulfides suffice. Biochemistry 2002, 41, 11488–11494. [Google Scholar] [CrossRef]
- Kabashima, T.; Yu, Z.Q.; Tang, C.H.; Nakagawa, Y.; Okumura, K.; Shibata, T.; Lu, J.Z.; Masaaki, K. A selective fluorescence reaction forpeptides and chromatographic analysis. Peptides 2008, 29, 356–363. [Google Scholar] [CrossRef]
- Fu, C.Y.; Xia, R.L.; Zhang, T.F.; Lu, Y.; Zhang, S.F.; Yu, Z.Q.; Jin, T.; Mou, X.Z. Hemokinin-1(4–11)-induced analgesia selectively up-regulates δ-opioid receptor expression in mice. PLoS One 2014, 9, e90446. [Google Scholar]
- Dubission, D.; Dennis, S.G. The formalin tests: A quantitative study of the analgesic effects of morphine, meperidine and brain stem stimulation in rats and cats. Pain 1977, 4, 161–174. [Google Scholar] [CrossRef]
- Malmberg, A.B.; Yaksh, T.L. Effect of continuous intrathecal infusion of u-conopeptides, N-type calcium channel blockers, on behavior and antinociception in the formalin and hot-plate tests in rats. Pain 1995, 60, 83–90. [Google Scholar] [CrossRef]
- Owoyele, V.B.; Oloriegbe, Y.Y.; Balogun, E.A.; Soladoye, A.O. Analgesic and anti-inflammatory properties of Nelsonia canescens leaf extract. J. Ethnopharmacol. 2005, 99, 153–156. [Google Scholar] [CrossRef]
- Rustay, N.R.; Wahlsten, D.; Crabbe, J.C. Influence of task parameters on rotarod performance and sensitivity to ethanol in mice. Behav. Brain Res. 2003, 141, 237–249. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, Y.; Tang, J.; Zhang, Y.; Xun, X.; Tang, D.; Peng, D.; Yi, J.; Liu, Z.; Shi, X. Synthesis and Analgesic Effects of μ-TRTX-Hhn1b on Models of Inflammatory and Neuropathic Pain. Toxins 2014, 6, 2363-2378. https://doi.org/10.3390/toxins6082363
Liu Y, Tang J, Zhang Y, Xun X, Tang D, Peng D, Yi J, Liu Z, Shi X. Synthesis and Analgesic Effects of μ-TRTX-Hhn1b on Models of Inflammatory and Neuropathic Pain. Toxins. 2014; 6(8):2363-2378. https://doi.org/10.3390/toxins6082363
Chicago/Turabian StyleLiu, Yu, Jianguang Tang, Yunxiao Zhang, Xiaohong Xun, Dongfang Tang, Dezheng Peng, Jianming Yi, Zhonghua Liu, and Xiaoliu Shi. 2014. "Synthesis and Analgesic Effects of μ-TRTX-Hhn1b on Models of Inflammatory and Neuropathic Pain" Toxins 6, no. 8: 2363-2378. https://doi.org/10.3390/toxins6082363
APA StyleLiu, Y., Tang, J., Zhang, Y., Xun, X., Tang, D., Peng, D., Yi, J., Liu, Z., & Shi, X. (2014). Synthesis and Analgesic Effects of μ-TRTX-Hhn1b on Models of Inflammatory and Neuropathic Pain. Toxins, 6(8), 2363-2378. https://doi.org/10.3390/toxins6082363