
G-Protein-Coupled Receptors: Next Generation Therapeutic Targets in Head and Neck Cancer?

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Galanin and Galanin Receptor Type 1 (GALR1)
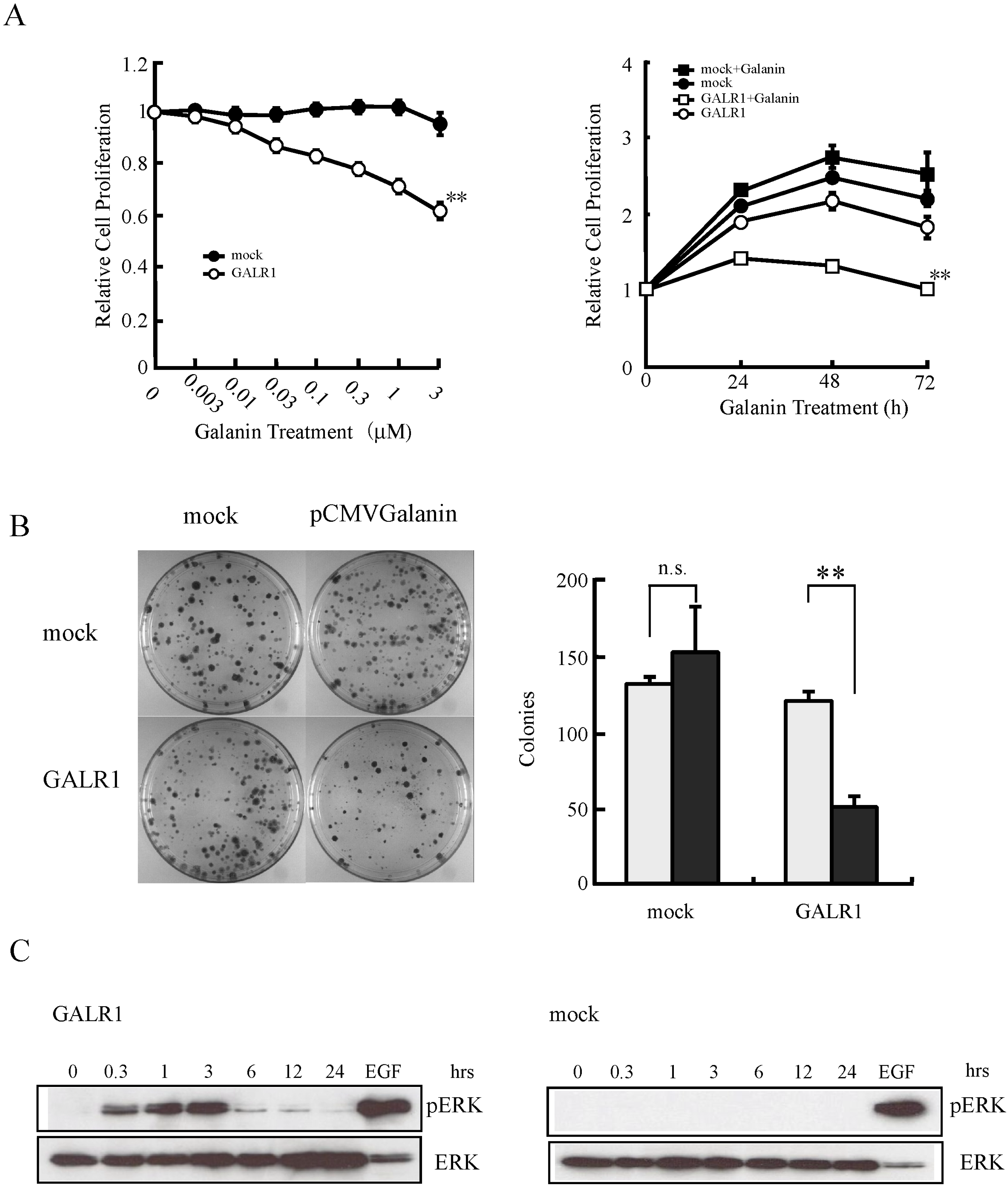
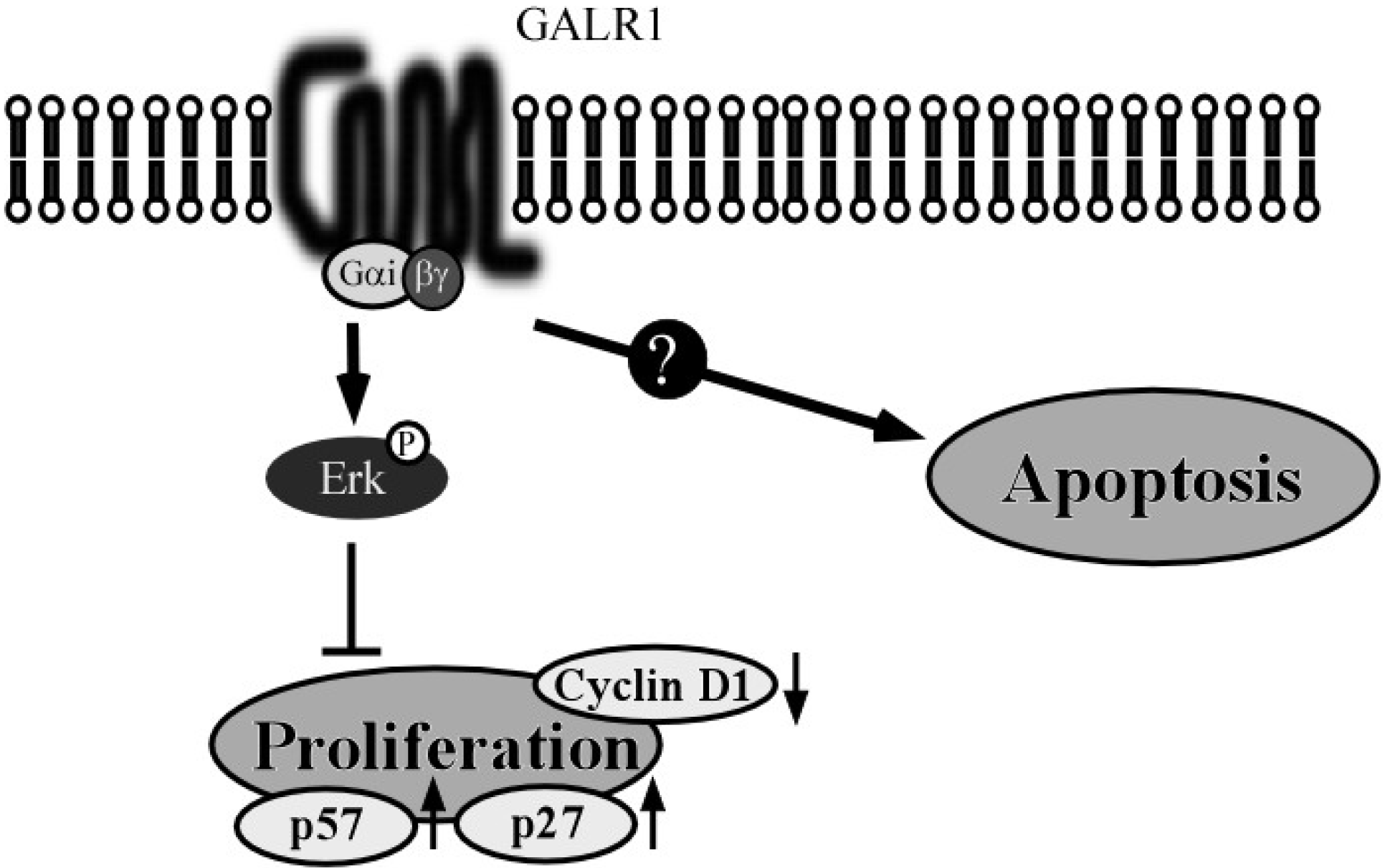
2.1. The GALR1 Signaling Pathway
2.2. GALR1 Function in HNSCC


2.3. Epigenetic Silencing of GALR1 in HNSCC and its Utility as a Prognostic Marker

3. Galanin and Galanin Receptor 2 (GALR2)
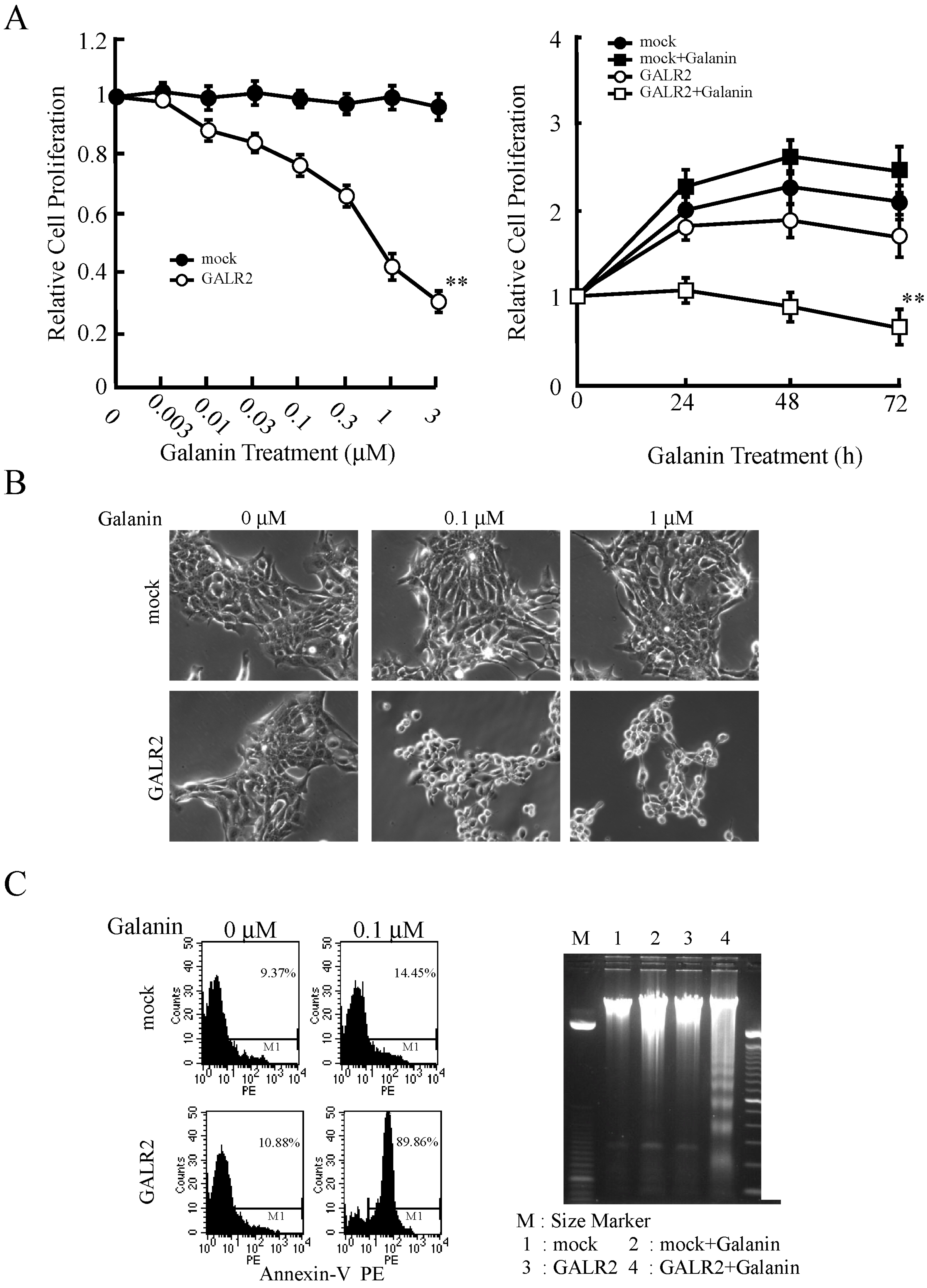
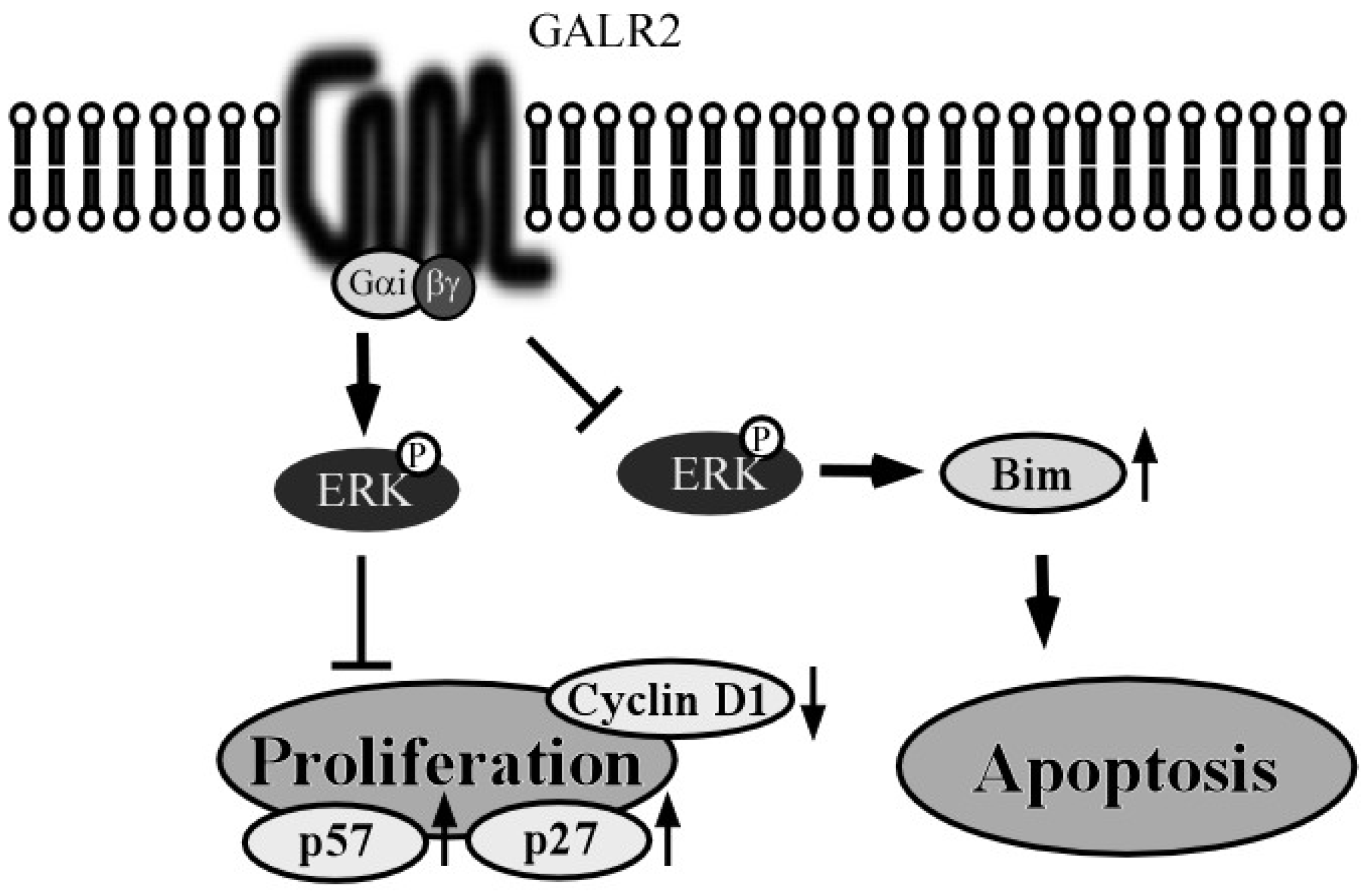
3.1. GALR2 Signaling Pathway
3.2. GALR2 Function in HNSCC


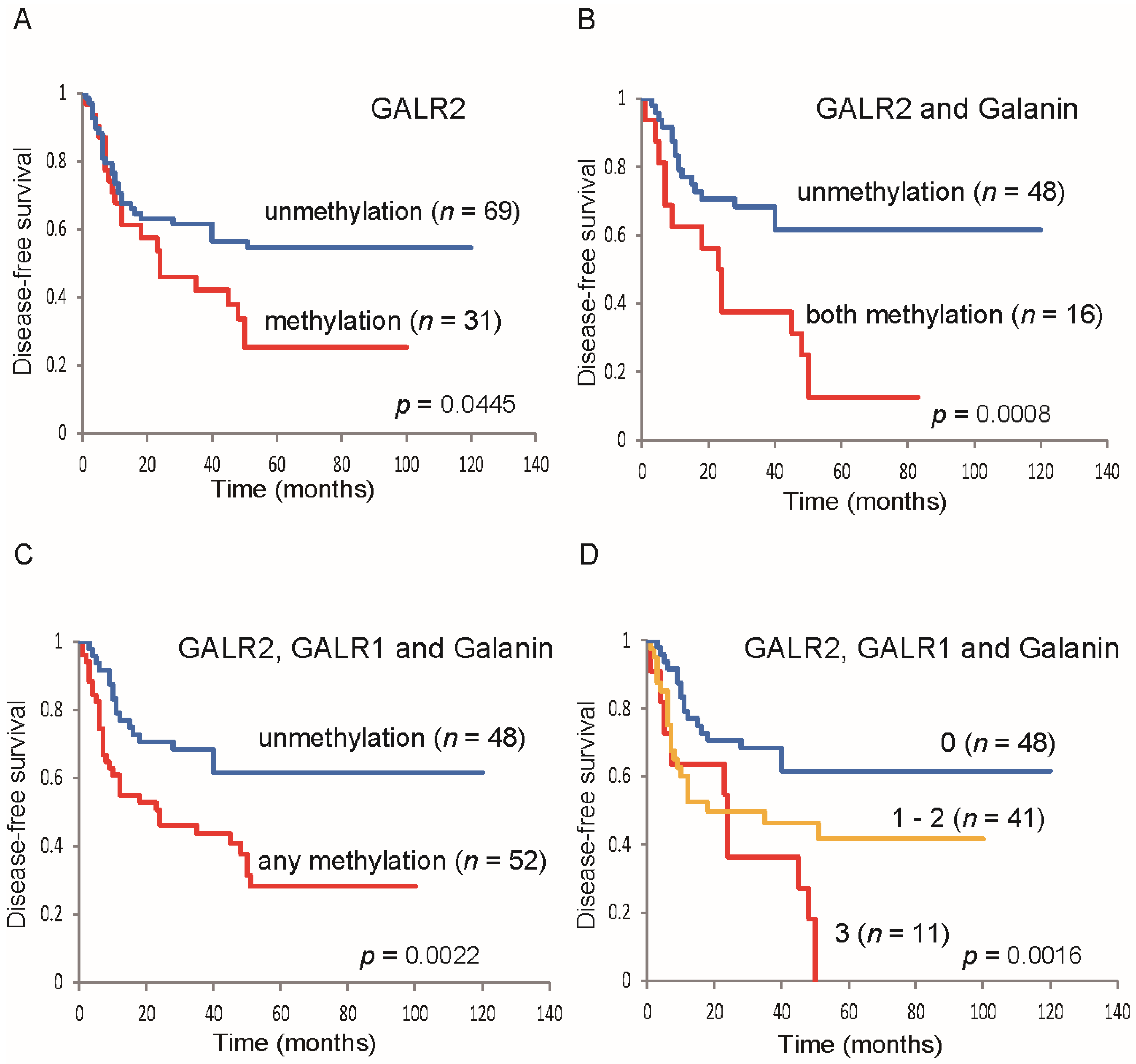
3.3. Epigenetic Silencing of GALR2 in HNSCC and its Utility as a Prognostic Marker

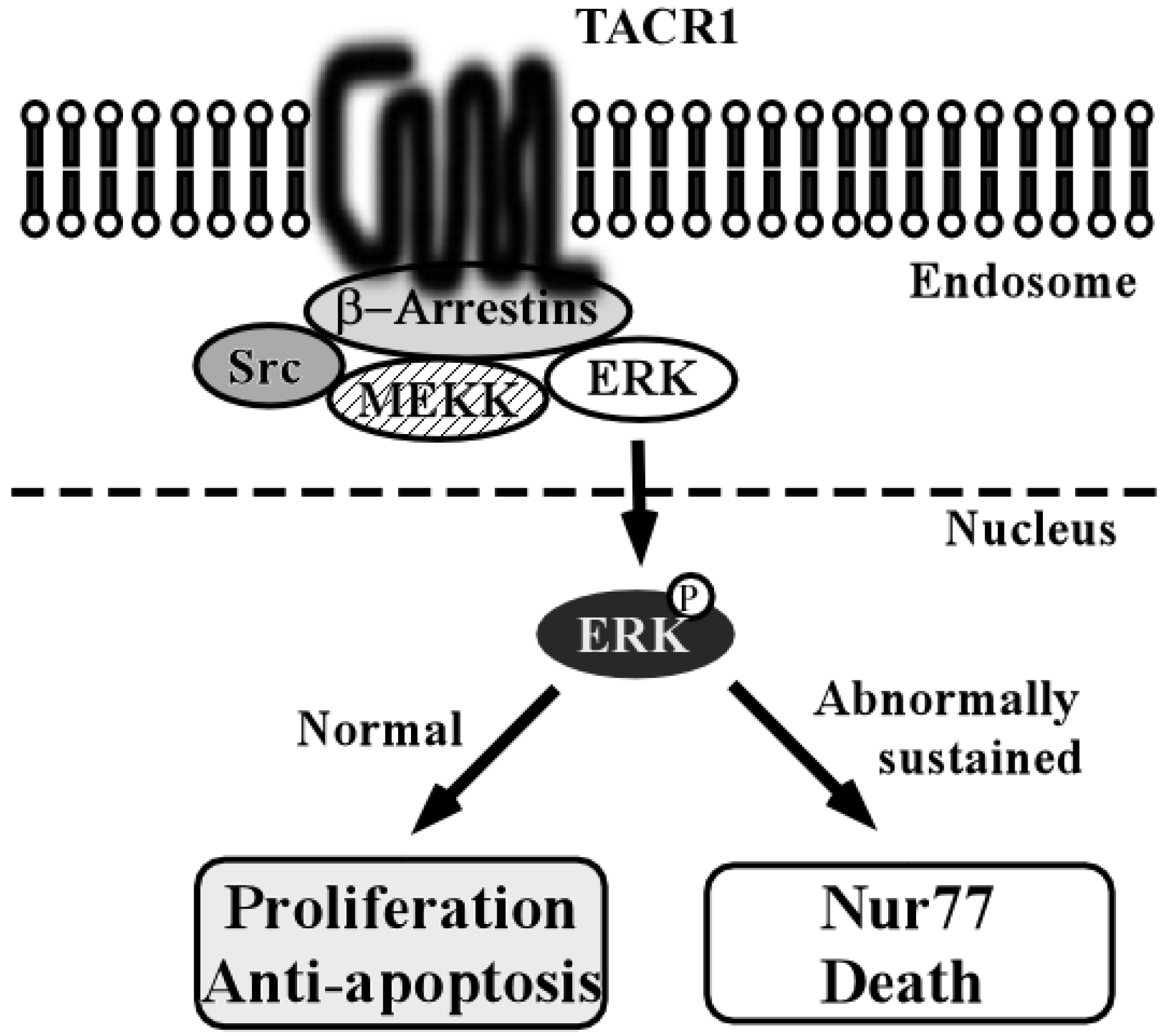
4. Tachykinin-1 and Tachykinin Receptor Type 1

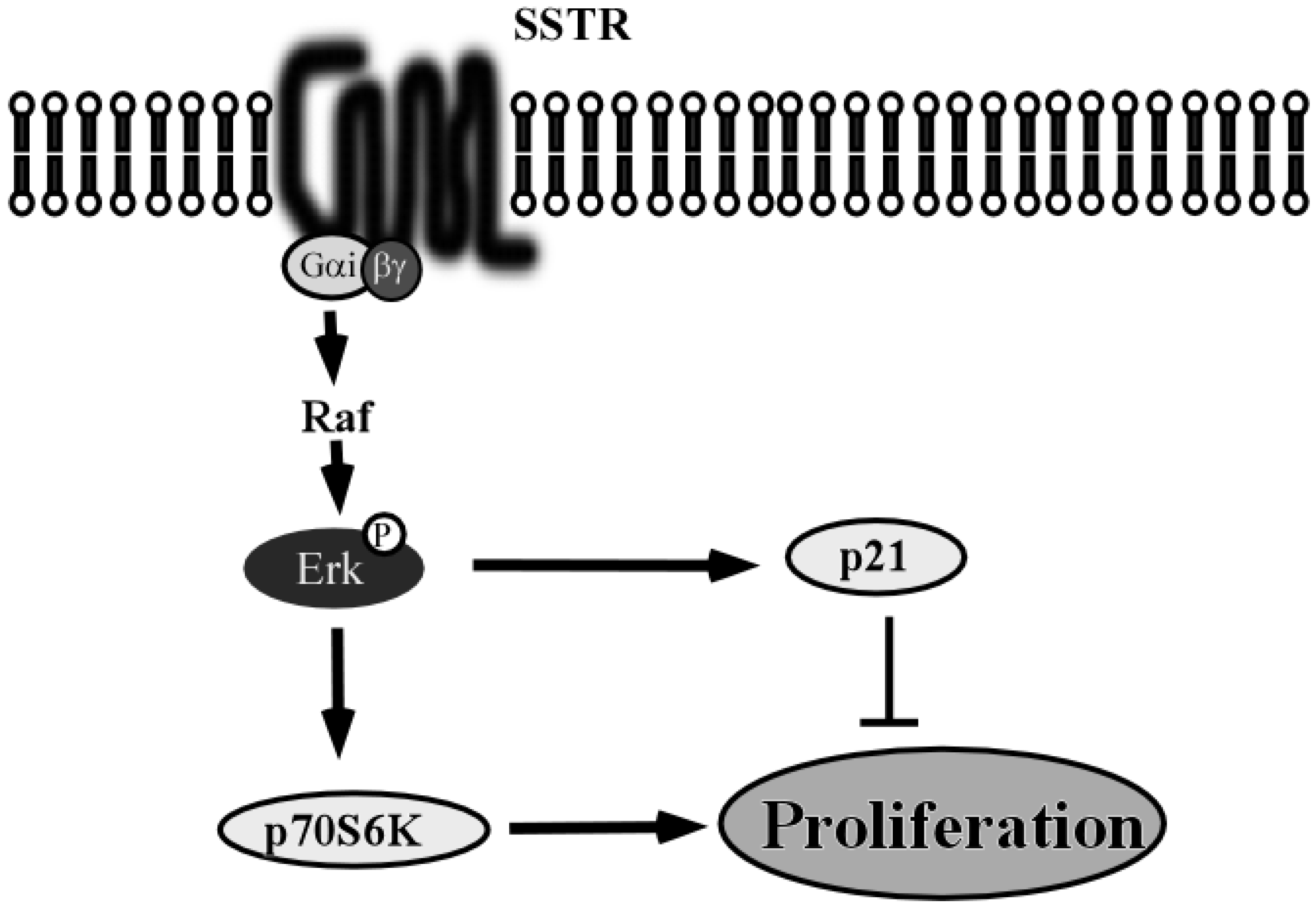
5. Somatostatin and Somatostatin Receptor 1

6. Future Directions for the Study of GPCRs in HNSCC
6.1. Loss of GPCR Signaling is a Prognostic Factor in HNSCC
6.2. GPCRs as Therapeutic Targets in HNSCC
6.3. Gene Therapy Using GPCRs
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Choong, N.; Vokes, E. Expanding role of the medical oncologist in the management of head and neck cancer. CA Cancer J. Clin. 2008, 58, 32–53. [Google Scholar] [CrossRef] [PubMed]
- Parfenov, M.; Pedamallu, C.S.; Gehlenborg, N.; Freeman, S.S.; Danilova, L.; Bristow, C.A.; Lee, S.; Hadjipanayis, A.G.; Ivanova, E.V.; Wilkerson, M.D.; et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15544–15549. [Google Scholar] [CrossRef] [PubMed]
- Matta, A.; Ralhan, R. Overview of current and future biologically based targeted therapies in head and neck squamous cell carcinoma. Head Neck Oncol. 2009, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.C.; Khuri, F.R.; Shin, D.M. Emerging drugs for head and neck cancer. Expert Opin. Emerg. Drugs 2004, 9, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Haddad, R.; Wirth, L.; Posner, M. Emerging drugs for head and neck cancer. Expert Opin. Emerg. Drugs 2006, 11, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Grandis, J.R. Emerging drugs for head and neck cancer. Expert Opin. Emerg. Drugs 2015, 20, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, T.; Hasegawa, Y.; Takahashi, S.; Monden, N.; Homma, A.; Okami, K.; Onozawa, Y.; Fujii, M.; Taguchi, T.; de Blas, B.; et al. Platinum-based chemotherapy plus cetuximab for the first-line treatment of Japanese patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck: Results of a phase II trial. Jpn. J. Clin. Oncol. 2013, 43, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Cordell, K.G.; D’Silva, N.; Prince, M.E.; Adams, M.E.; Fisher, S.G.; Wolf, G.T.; Carey, T.E.; Bradford, C.R. Expression of p53 and Bcl-xL as predictive markers for larynx preservation in advanced laryngeal cancer. Arch. Otolaryngol. Head Neck Surg. 2008, 134, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Bradford, C.R.; Kumar, B.; Bellile, E.; Lee, J.; Taylor, J.; D’Silva, N.; Cordell, K.; Kleer, C.; Kupfer, R.; Kumar, P.; et al. Biomarkers in advanced larynx cancer. Laryngoscope 2014, 124, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Hasegawa, M.; Yamashita, Y.; Matayoshi, S.; Kiyuna, A.; Agena, S.; Uehara, T.; Maeda, H.; Suzuki, M. Prognostic value of human papillomavirus and squamous cell carcinoma antigen in head and neck squamous cell carcinoma. Cancer Sci. 2012, 103, 2127–2134. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Thomas, S.M.; Freilino, M.; Joyce, S.; Sahu, A.; Maxwell, J.; Argiris, A.; Seethala, R.; Grandis, J.R. Targeting GPCR-mediated p70S6K activity may improve head and neck cancer response to cetuximab. Clin. Cancer Res. 2011, 17, 4996–5004. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Freilino, M.L.; Joyce, S.C.; Sen, M.; Thomas, S.M.; Sahu, A.; Cassell, A.; Chen, C.S.; Grandis, J.R. Antitumor mechanisms of targeting the PDK1 pathway in head and neck cancer. Mol. Cancer Ther. 2012, 11, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Habert-Ortoli, E.; Amiranoff, B.; Loquet, I.; Laburthe, M.; Mayaux, J.F. Molecular cloning of a functional human galanin receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 9780–9783. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Hashemi, T.; Fried, S.; Clemmons, A.L.; Hawes, B.E. Differential intracellular signaling of the GalR1 and GalR2 galanin receptor subtypes. Biochemistry 1998, 37, 6711–6717. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.E.; Walker, M.W.; Artymyshyn, R.; Bard, J.; Borowsky, B.; Tamm, J.A.; Yao, W.J.; Vaysse, P.J.; Branchek, T.A.; Gerald, C.; et al. Cloned human and rat galanin GALR3 receptors. Pharmacology and activation of G-protein inwardly rectifying K+ channels. J. Biol. Chem. 1998, 273, 23321–23326. [Google Scholar] [CrossRef] [PubMed]
- Friday, B.B.; Adjei, A.A. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin. Cancer Res. 2008, 14, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Henson, B.S.; Neubig, R.R.; Jang, I.; Ogawa, T.; Zhang, Z.; Carey, T.E.; D’Silva, N.J. Galanin receptor 1 has anti-proliferative effects in oral squamous cell carcinoma. J. Biol. Chem. 2005, 280, 22564–22571. [Google Scholar] [CrossRef] [PubMed]
- Gutkind, J.S. The pathways connecting G protein-coupled receptors to the nucleus through divergent mitogen-activated protein kinase cascades. J. Biol. Chem. 1998, 273, 1839–1842. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, S.; Ogawa, T.; Jung, K.Y.; Muallem, A.; Mineta, H.; Fisher, S.G.; Grenman, R.; Carey, T.E. Identification of new minimally lost regions on 18q in head and neck squamous cell carcinoma. Cancer Res. 2000, 60, 3397–3403. [Google Scholar] [PubMed]
- Kanazawa, T.; Iwashita, T.; Kommareddi, P.; Nair, T.; Misawa, K.; Misawa, Y.; Ueda, Y.; Tono, T.; Carey, T.E. Galanin and galanin receptor type 1 suppress proliferation in squamous carcinoma cells: Activation of the extracellular signal regulated kinase pathway and induction of cyclin-dependent kinase inhibitors. Oncogene 2007, 26, 5762–5771. [Google Scholar] [CrossRef] [PubMed]
- Pumiglia, K.M.; Decker, S.J. Cell cycle arrest mediated by the MEK/mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci. USA 1997, 94, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Dixon, B.S.; Evanoff, D.; Fang, W.B.; Dennis, M.J. Bradykinin B1 receptor blocks PDGF-induced mitogenesis by prolonging ERK activation and increasing p27Kip1. Am. J. Physiol. Cell Physiol. 2002, 283, C193–C203. [Google Scholar] [CrossRef] [PubMed]
- Lahlou, H.; Saint-Laurent, N.; Esteve, J.P.; Eychene, A.; Pradayrol, L.; Pyronnet, S.; Susini, C. sst2 Somatostatin receptor inhibits cell proliferation through Ras-, Rap1-, and B-Raf-dependent ERK2 activation. J. Biol. Chem. 2003, 278, 39356–39371. [Google Scholar] [CrossRef] [PubMed]
- Woods, D.; Parry, D.; Cherwinski, H.; Bosch, E.; Lees, E.; McMahon, M. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol. Cell. Biol. 1997, 17, 5598–5611. [Google Scholar] [PubMed]
- Gendron, L.; Oligny, J.F.; Payet, M.D.; Gallo-Payet, N. Cyclic AMP-independent involvement of Rap1/B-Raf in the angiotensin II AT2 receptor signaling pathway in NG108-15 cells. J. Biol. Chem. 2003, 278, 3606–3614. [Google Scholar] [CrossRef] [PubMed]
- Kranenburg, O.; Moolenaar, W.H. Ras-MAP kinase signaling by lysophosphatidic acid and other G protein-coupled receptor agonists. Oncogene 2001, 20, 1540–1546. [Google Scholar] [CrossRef] [PubMed]
- Esposito, V.; Baldi, A.; de Luca, A.; Groger, A.M.; Loda, M.; Giordano, G.G.; Caputi, M.; Baldi, F.; Pagano, M.; Giordano, A. Prognostic role of the cyclin-dependent kinase inhibitor p27 in non-small cell lung cancer. Cancer Res. 1997, 57, 3381–3385. [Google Scholar] [PubMed]
- Masuda, T.A.; Inoue, H.; Sonoda, H.; Mine, S.; Yoshikawa, Y.; Nakayama, K.; Nakayama, K.; Mori, M. Clinical and biological significance of S-phase kinase-associated protein 2 (Skp2) Gene expression in gastric carcinoma: Modulation of malignant phenotype by Skp2 overexpression, possibly via p27 proteolysis. Cancer Res. 2002, 62, 3819–3825. [Google Scholar] [PubMed]
- Massarelli, E.; Brown, E.; Tran, N.K.; Liu, D.D.; Izzo, J.G.; Lee, J.J.; El-Naggar, A.K.; Hong, W.K.; Papadimitrakopoulou, V.A. Loss of E-cadherin and p27 expression is associated with head and neck squamous tumorigenesis. Cancer 2005, 103, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.J.; Florl, A.R.; Seifert, H.H.; Schulz, W.A. Multiple mechanisms downregulate CDKN1C in human bladder cancer. Int. J. Cancer 2005, 114, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.; Amos, C.I.; Luthra, R.; Lynch, P.M.; Levin, B.; Frazier, M.L. Effects of cyclin D1 polymorphism on age of onset of hereditary nonpolyposis colorectal cancer. Cancer Res. 2000, 60, 249–252. [Google Scholar] [PubMed]
- Akervall, J.; Bockmuhl, U.; Petersen, I.; Yang, K.; Carey, T.E.; Kurnit, D.M. The gene ratios c-MYC:cyclin-dependent kinase (CDK)N2A and CCND1:CDKN2A correlate with poor prognosis in squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2003, 9, 1750–1755. [Google Scholar] [PubMed]
- Nancarrow, D.J.; Handoko, H.Y.; Smithers, B.M.; Gotley, D.C.; Drew, P.A.; Watson, D.I.; Clouston, A.D.; Hayward, N.K.; Whiteman, D.C. Genome-wide copy number analysis in esophageal adenocarcinoma using high-density single-nucleotide polymorphism arrays. Cancer Res. 2008, 68, 4163–4172. [Google Scholar] [CrossRef] [PubMed]
- Doufekas, K.; Hadwin, R.; Kandimalla, R.; Jones, A.; Mould, T.; Crowe, S.; Olaitan, A.; Macdonald, N.; Fiegl, H.; Wik, E.; et al. GALR1 methylation in vaginal swabs is highly accurate in identifying women with endometrial cancer. Int. J. Gynecol. Cancer 2013, 23, 1050–1055. [Google Scholar] [CrossRef] [PubMed]
- Jee, K.J.; Persson, M.; Heikinheimo, K.; Passador-Santos, F.; Aro, K.; Knuutila, S.; Odell, E.W.; Makitie, A.; Sundelin, K.; Stenman, G.; et al. Genomic profiles and CRTC1-MAML2 fusion distinguish different subtypes of mucoepidermoid carcinoma. Mod. Pathol. 2013, 26, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Srivastava, S. Epigenetics in cancer: Implications for early detection and prevention. Lancet Oncol. 2002, 3, 755–763. [Google Scholar] [CrossRef]
- Misawa, K.; Ueda, Y.; Kanazawa, T.; Misawa, Y.; Jang, I.; Brenner, J.C.; Ogawa, T.; Takebayashi, S.; Grenman, R.A.; Herman, J.G.; et al. Epigenetic inactivation of galanin receptor 1 in head and neck cancer. Clin. Cancer Res. 2008, 14, 7604–7613. [Google Scholar] [CrossRef] [PubMed]
- Misawa, K.; Kanazawa, T.; Misawa, Y.; Uehara, T.; Imai, A.; Takahashi, G.; Takebayashi, S.; Cole, A.; Carey, T.E.; Mineta, H. Galanin has tumor suppressor activity and is frequently inactivated by aberrant promoter methylation in head and neck cancer. Transl. Oncol. 2013, 6, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.; Gundlach, A.L.; Kofler, B. The galanin peptide family: Receptor pharmacology, pleiotropic biological actions, and implications in health and disease. Pharmacol. Ther. 2007, 115, 177–207. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.E.; Forray, C.; Walker, M.W.; Jones, K.A.; Tamm, J.A.; Bard, J.; Branchek, T.A.; Linemeyer, D.L.; Gerald, C. Expression cloning of a rat hypothalamic galanin receptor coupled to phosphoinositide turnover. J. Biol. Chem. 1997, 272, 24612–24616. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Clemmons, A.; Strader, C.; Bayne, M. Evidence for hydrophobic interaction between galanin and the GalR1 galanin receptor and GalR1-mediated ligand internalization: Fluorescent probing with a fluorescein-galanin. Biochemistry 1998, 37, 9528–9535. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, T.; Misawa, K.; Carey, T.E. Galanin receptor subtypes 1 and 2 as therapeutic targets in head and neck squamous cell carcinoma. Expert Opin. Ther. Targets 2010, 14, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, T.; Misawa, K.; Misawa, Y.; Maruta, M.; Uehara, T.; Kawada, K.; Nagatomo, T.; Ichimura, K. Galanin receptor 2 utilizes distinct signaling pathways to suppress cell proliferation and induce apoptosis in HNSCC. Mol. Med. Rep. 2014, 10, 1289–1294. [Google Scholar] [CrossRef] [PubMed]
- Fathi, Z.; Battaglino, P.M.; Iben, L.G.; Li, H.; Baker, E.; Zhang, D.; McGovern, R.; Mahle, C.D.; Sutherland, G.R.; Iismaa, T.P.; et al. Molecular characterization, pharmacological properties and chromosomal localization of the human GALR2 galanin receptor. Brain Res. Mol. Brain Res. 1998, 58, 156–169. [Google Scholar] [CrossRef]
- Wang, S.; Hashemi, T.; He, C.; Strader, C.; Bayne, M. Molecular cloning and pharmacological characterization of a new galanin receptor subtype. Mol. Pharmacol. 1997, 52, 337–343. [Google Scholar] [PubMed]
- Hobson, S.A.; Holmes, F.E.; Kerr, N.C.; Pope, R.J.; Wynick, D. Mice deficient for galanin receptor 2 have decreased neurite outgrowth from adult sensory neurons and impaired pain-like behaviour. J. Neurochem. 2006, 99, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Elliott-Hunt, C.R.; Pope, R.J.; Vanderplank, P.; Wynick, D. Activation of the galanin receptor 2 (GalR2) protects the hippocampus from neuronal damage. J. Neurochem. 2007, 100, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Wittau, N.; Grosse, R.; Kalkbrenner, F.; Gohla, A.; Schultz, G.; Gudermann, T. The galanin receptor type 2 initiates multiple signaling pathways in small cell lung cancer cells by coupling to G(q), G(i) and G(12) proteins. Oncogene 2000, 19, 4199–4209. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Lang, R.; Moritz, K.; Santic, R.; Hermann, A.; Sperl, W.; Kofler, B. Galanin receptor subtype GalR2 mediates apoptosis in SH-SY5Y neuroblastoma cells. Endocrinology 2004, 145, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Tofighi, R.; Joseph, B.; Xia, S.; Xu, Z.Q.; Hamberger, B.; Hokfelt, T.; Ceccatelli, S. Galanin decreases proliferation of PC12 cells and induces apoptosis via its subtype 2 receptor (GalR2). Proc. Natl. Acad. Sci. USA 2008, 105, 2717–2722. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, T.; Kommareddi, P.K.; Iwashita, T.; Kumar, B.; Misawa, K.; Misawa, Y.; Jang, I.; Nair, T.S.; Iino, Y.; Carey, T.E. Galanin receptor subtype 2 suppresses cell proliferation and induces apoptosis in p53 mutant head and neck cancer cells. Clin. Cancer Res. 2009, 15, 2222–2230. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Kanazawa, T.; Mizukami, H.; Uchibori, R.; Tsukahara, T.; Urabe, M.; Kume, A.; Misawa, K.; Carey, T.E.; Suzuki, M.; et al. Novel anti-tumor mechanism of galanin receptor type 2 in head and neck squamous cell carcinoma cells. Cancer Sci. 2014, 105, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, T.; Seki, N.; Shimizu, S.; Kikkawa, N.; Tsukada, J.; Shimada, H.; Sasaki, K.; Hanazawa, T.; Okamoto, Y.; Hata, A. The galanin signaling cascade is a candidate pathway regulating oncogenesis in human squamous cell carcinoma. Genes Chromosomes Cancer 2009, 48, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Henson, B.S.; Russo, N.; Tsodikov, A.; D’Silva, N.J. Rap1 mediates galanin receptor 2-induced proliferation and survival in squamous cell carcinoma. Cell Signal. 2011, 23, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; van Tubergen, E.A.; Scanlon, C.S.; Vander Broek, R.; Lints, J.P.; Liu, M.; Russo, N.; Inglehart, R.C.; Wang, Y.; Polverini, P.J.; et al. The G protein-coupled receptor GALR2 promotes angiogenesis in head and neck cancer. Mol. Cancer Ther. 2014, 13, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Pin, J.P.; Neubig, R.; Bouvier, M.; Devi, L.; Filizola, M.; Javitch, J.A.; Lohse, M.J.; Milligan, G.; Palczewski, K.; Parmentier, M.; et al. International Union of Basic and Clinical Pharmacology. LXVII. Recommendations for the recognition and nomenclature of G protein-coupled receptor heteromultimers. Pharmacol. Rev. 2007, 59, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.; Kwabi-Addo, B.; Ittmann, M.; Jelinek, J.; Shen, L.; Yu, Y.; Issa, J.P. Identification of novel tumor markers in prostate, colon and breast cancer by unbiased methylation profiling. PLoS ONE 2008, 3, e2079. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, H.Y.; Ma, Z.Z.; Lu, W.; Wang, Y.F.; Zhu, J.D. Methylation profiling of twenty four genes and the concordant methylation behaviours of nineteen genes that may contribute to hepatocellular carcinogenesis. Cell Res. 2003, 13, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.C.; Lee, H.C.; Cho, D.H.; Choi, E.Y.; Cho, Y.K.; Ha, Y.J.; Choi, P.W.; Roh, S.A.; Kim, S.Y.; Kim, Y.S. Genome-wide identification of possible methylation markers chemosensitive to targeted regimens in colorectal cancers. J. Cancer Res. Clin. Oncol. 2011, 137, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Misawa, Y.; Misawa, K.; Kanazawa, T.; Uehara, T.; Endo, S.; Mochizuki, D.; Yamatodani, T.; Carey, T.E.; Mineta, H. Tumor suppressor activity and inactivation of galanin receptor type 2 by aberrant promoter methylation in head and neck cancer. Cancer 2014, 120, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Pennefather, J.N.; Lecci, A.; Candenas, M.L.; Patak, E.; Pinto, F.M.; Maggi, C.A. Tachykinins and tachykinin receptors: A growing family. Life Sci. 2004, 74, 1445–1463. [Google Scholar] [CrossRef] [PubMed]
- Pinto, F.M.; Almeida, T.A.; Hernandez, M.; Devillier, P.; Advenier, C.; Candenas, M.L. mRNA expression of tachykinins and tachykinin receptors in different human tissues. Eur. J. Pharmacol. 2004, 494, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Jaafari, N.; Hua, G.; Adelaide, J.; Jule, Y.; Imbert, J. Expression of the tachykinin receptor mRNAs in healthy human colon. Eur. J. Pharmacol. 2008, 599, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Severini, C.; Improta, G.; Falconieri-Erspamer, G.; Salvadori, S.; Erspamer, V. The tachykinin peptide family. Pharmacol. Rev. 2002, 54, 285–322. [Google Scholar] [CrossRef] [PubMed]
- Koon, H.W.; Zhao, D.; Na, X.; Moyer, M.P.; Pothoulakis, C. Metalloproteinases and transforming growth factor-alpha mediate substance P-induced mitogen-activated protein kinase activation and proliferation in human colonocytes. J. Biol. Chem. 2004, 279, 45519–45527. [Google Scholar] [CrossRef] [PubMed]
- Lieb, K.; Fiebich, B.L.; Berger, M.; Bauer, J.; Schulze-Osthoff, K. The neuropeptide substance P activates transcription factor NF-kappa B and kappa B-dependent gene expression in human astrocytoma cells. J. Immunol. 1997, 159, 4952–4958. [Google Scholar] [PubMed]
- Rameshwar, P.; Gascon, P. Induction of negative hematopoietic regulators by neurokinin-A in bone marrow stroma. Blood 1996, 88, 98–106. [Google Scholar] [PubMed]
- Rosso, M.; Munoz, M.; Berger, M. The role of neurokinin-1 receptor in the microenvironment of inflammation and cancer. ScientificWorldJournal 2012, 2012, 381434. [Google Scholar] [PubMed]
- Steinhoff, M.S.; von Mentzer, B.; Geppetti, P.; Pothoulakis, C.; Bunnett, N.W. Tachykinins and their receptors: Contributions to physiological control and the mechanisms of disease. Physiol. Rev. 2014, 94, 265–301. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Olaru, A.; Yang, J.; Sato, F.; Cheng, Y.; Kan, T.; Mori, Y.; Mantzur, C.; Paun, B.; Hamilton, J.P.; et al. Hypermethylation of tachykinin-1 is a potential biomarker in human esophageal cancer. Clin. Cancer Res. 2007, 13, 6293–6300. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Cai, K.; Cheng, Y.; Wang, S.; Paun, B.; Hamilton, J.P.; Jin, Z.; Sato, F.; Berki, A.T.; Kan, T.; et al. A genome-wide search identifies epigenetic silencing of somatostatin, tachykinin-1, and 5 other genes in colon cancer. Gastroenterology 2006, 131, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, J.; van Neste, L.; Glockner, S.C.; Dhir, M.; Calmon, M.F.; Deregowski, V.; van Criekinge, W.; Vlassenbroeck, I.; Koch, A.; Chan, T.A.; et al. Biomarkers for detection and prognosis of breast cancer identified by a functional hypermethylome screen. Epigenetics 2012, 7, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Misawa, K.; Kanazawa, T.; Misawa, Y.; Imai, A.; Uehara, T.; Mochizuki, D.; Endo, S.; Takahashi, G.; Mineta, H. Frequent promoter hypermethylation of tachykinin-1 and tachykinin receptor type 1 is a potential biomarker for head and neck cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Mori, Y.; Hamilton, J.P.; Olaru, A.; Sato, F.; Yang, J.; Ito, T.; Kan, T.; Agarwal, R.; Meltzer, S.J. Hypermethylation of the somatostatin promoter is a common, early event in human esophageal carcinogenesis. Cancer 2008, 112, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Laissue, J.A. Multiple actions of somatostatin in neoplastic disease. Trends Pharmacol. Sci. 1995, 16, 110–115. [Google Scholar] [CrossRef]
- Zhao, J.; Liang, Q.; Cheung, K.F.; Kang, W.; Dong, Y.; Lung, R.W.; Tong, J.H.; To, K.F.; Sung, J.J.; Yu, J. Somatostatin receptor 1, a novel EBV-associated CpG hypermethylated gene, contributes to the pathogenesis of EBV-associated gastric cancer. Br. J. Cancer 2013, 108, 2557–2564. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.; Soutto, M.; Peng, D.; Hu, T.; Marshal, D.; El-Rifai, W. Epigenetic silencing of somatostatin in gastric cancer. Dig. Dis. Sci. 2011, 56, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Watt, H.L.; Rachid, Z.; Jean-Claude, B.J. The Concept of Divergent Targeting through the Activation and Inhibition of Receptors as a Novel Chemotherapeutic Strategy: Signaling Responses to Strong DNA-Reactive Combinatorial Mimicries. J. Signal Transduct. 2012, 2012, 282050. [Google Scholar] [CrossRef] [PubMed]
- Misawa, K.; Misawa, Y.; Kondo, H.; Mochizuki, D.; Imai, A.; Fukushima, H.; Uehara, T.; Kanazawa, T.; Mineta, H. Aberrant methylation inactivates somatostatin and somatostatin receptor type 1 in head and neck squamous cell carcinoma. PLoS ONE 2015, 10, e0118588. [Google Scholar] [CrossRef] [PubMed]
- Toyooka, S.; Maruyama, R.; Toyooka, K.O.; McLerran, D.; Feng, Z.; Fukuyama, Y.; Virmani, A.K.; Zochbauer-Muller, S.; Tsukuda, K.; Sugio, K.; et al. Smoke exposure, histologic type and geography-related differences in the methylation profiles of non-small cell lung cancer. Int. J. Cancer 2003, 103, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Berman, D.; Lu, C.; Wistuba, I.I.; Roth, J.A.; Frazier, M.; Spitz, M.R.; Wu, X. Aberrant promoter methylation profile and association with survival in patients with non-small cell lung cancer. Clin. Cancer Res. 2006, 12, 7329–7338. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Cordell, K.G.; Lee, J.S.; Worden, F.P.; Prince, M.E.; Tran, H.H.; Wolf, G.T.; Urba, S.G.; Chepeha, D.B.; Teknos, T.N.; et al. EGFR, p16, HPV Titer, Bcl-xL and p53, sex, and smoking as indicators of response to therapy and survival in oropharyngeal cancer. J. Clin. Oncol. 2008, 26, 3128–3137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jeltsch, A. The application of next generation sequencing in DNA methylation analysis. Genes (Basel) 2010, 1, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Iishi, H.; Tatsuta, M.; Baba, M.; Uehara, H.; Yano, H.; Nakaizumi, A. Chemoprevention by galanin against colon carcinogenesis induced by azoxymethane in Wistar rats. Int. J. Cancer 1995, 61, 861–863. [Google Scholar] [CrossRef] [PubMed]
- Iishi, H.; Tatsuta, M.; Baba, M.; Yano, H.; Iseki, K.; Uehara, H.; Nakaizumi, A. Inhibition by galanin of experimental carcinogenesis induced by azaserine in rat pancreas. Int. J. Cancer 1998, 75, 396–399. [Google Scholar] [CrossRef]
- El-Salhy, M.; Tjomsland, V.; Theodorsson, E. Effects of triple treatment with octreotide, galanin and serotonin on a human pancreas cancer cell line in xenografts. Histol. Histopathol. 2005, 20, 745–752. [Google Scholar] [PubMed]
- Merino, O.R.; Lindberg, R.D.; Fletcher, G.H. An analysis of distant metastases from squamous cell carcinoma of the upper respiratory and digestive tracts. Cancer 1977, 40, 145–151. [Google Scholar] [CrossRef]
- Yoo, G.H.; Moon, J.; Leblanc, M.; Lonardo, F.; Urba, S.; Kim, H.; Hanna, E.; Tsue, T.; Valentino, J.; Ensley, J.; et al. A phase 2 trial of surgery with perioperative INGN 201 (Ad5CMV-p53) gene therapy followed by chemoradiotherapy for advanced, resectable squamous cell carcinoma of the oral cavity, oropharynx, hypopharynx, and larynx: Report of the Southwest Oncology Group. Arch. Otolaryngol. Head Neck Surg. 2009, 135, 869–874. [Google Scholar] [PubMed]
- Moss, R.B.; Milla, C.; Colombo, J.; Accurso, F.; Zeitlin, P.L.; Clancy, J.P.; Spencer, L.T.; Pilewski, J.; Waltz, D.A.; Dorkin, H.L.; et al. Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: A randomized placebo-controlled phase 2B trial. Hum. Gene Ther. 2007, 18, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, A.; Mimuro, J.; Mizukami, H.; Kashiwakura, Y.; Takano, K.; Ohmori, T.; Madoiwa, S.; Ozawa, K.; Sakata, Y. Liver-restricted expression of the canine factor VIII gene facilitates prevention of inhibitor formation in factor VIII-deficient mice. J. Gene Med. 2009, 11, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, K.; Fan, D.S.; Shen, Y.; Muramatsu, S.; Fujimoto, K.; Ikeguchi, K.; Ogawa, M.; Urabe, M.; Kume, A.; Nakano, I. Gene therapy of Parkinson’s disease using adeno-associated virus (AAV) vectors. J. Neural Transm. Suppl. 2000, 7, 181–191. [Google Scholar]
- Kanazawa, T.; Mizukami, H.; Okada, T.; Hanazono, Y.; Kume, A.; Nishino, H.; Takeuchi, K.; Kitamura, K.; Ichimura, K.; Ozawa, K. Suicide gene therapy using AAV-HSVtk/ganciclovir in combination with irradiation results in regression of human head and neck cancer xenografts in nude mice. Gene Ther. 2003, 10, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, T.; Mizukami, H.; Nishino, H.; Okada, T.; Hanazono, Y.; Kume, A.; Kitamura, K.; Ichimura, K.; Ozawa, K. Topoisomerase inhibitors enhance the cytocidal effect of AAV-HSVtk/ganciclovir on head and neck cancer cells. Int. J. Oncol. 2004, 25, 729–735. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanazawa, T.; Misawa, K.; Misawa, Y.; Uehara, T.; Fukushima, H.; Kusaka, G.; Maruta, M.; Carey, T.E. G-Protein-Coupled Receptors: Next Generation Therapeutic Targets in Head and Neck Cancer? Toxins 2015, 7, 2959-2984. https://doi.org/10.3390/toxins7082959
Kanazawa T, Misawa K, Misawa Y, Uehara T, Fukushima H, Kusaka G, Maruta M, Carey TE. G-Protein-Coupled Receptors: Next Generation Therapeutic Targets in Head and Neck Cancer? Toxins. 2015; 7(8):2959-2984. https://doi.org/10.3390/toxins7082959
Chicago/Turabian StyleKanazawa, Takeharu, Kiyoshi Misawa, Yuki Misawa, Takayuki Uehara, Hirofumi Fukushima, Gen Kusaka, Mikiko Maruta, and Thomas E. Carey. 2015. "G-Protein-Coupled Receptors: Next Generation Therapeutic Targets in Head and Neck Cancer?" Toxins 7, no. 8: 2959-2984. https://doi.org/10.3390/toxins7082959
APA StyleKanazawa, T., Misawa, K., Misawa, Y., Uehara, T., Fukushima, H., Kusaka, G., Maruta, M., & Carey, T. E. (2015). G-Protein-Coupled Receptors: Next Generation Therapeutic Targets in Head and Neck Cancer? Toxins, 7(8), 2959-2984. https://doi.org/10.3390/toxins7082959